14.6.2026
Aktualizace ČERVEN 2026
Aktualizace AISLP červen 2026 je připravena ke stažení.
Vážení uživatelé programu AISLP,
přinášíme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální červnové verzi AISLP.
Convulex 300 mg/ml perorální roztok a 150 mg, 300 mg a 500 mg enterosolventní měkké tobolky (kyselina valproová) – antiepileptikum – zrušení indikace profylaxe migrény.
Hetronifly 10 mg/ml koncentrát pro infuzní roztok (serplulimab) – cytostatikum, monoklonální protilátka – rozšíření indikací, nově: v kombinaci s karboplatinou a pemetrexedem k první linii léčby dospělých pacientů s neskvamózním nemalobuněčným karcinomem plic (NSCLC) bez pozitivních mutací EGFR, ALK nebo ROS1, kteří mají lokálně pokročilý NSCLC a kteří nejsou vhodnými kandidáty na chirurgickou léčbu či radioterapii, nebo metastazující NSCLC; v kombinaci s chemoterapií na bázi fluorpyrimidinu a platiny k první linii léčby dospělých pacientů s neresekovatelným, lokálně pokročilým, recidivujícím nebo metastazujícím spinocelulárním karcinomem jícnu, jejichž nádory exprimují PD-L1 s CPS 5 a více.
Imfinzi 50 mg/ml koncentrát pro infuzní roztok (durvalumab) – cytostatikum, monoklonální protilátka – rozšíření indikací, nově: v kombinaci s chemoterapií FLOT jako neoadjuvantní a adjuvantní léčba, následovaná adjuvantní monoterapií přípravkem Imfinzi, k léčbě dospělých pacientů s resekovatelným adenokarcinomem žaludku nebo gastroezofageální junkce.
Imcivree 10 mg/ml injekční roztok (setmelanotid) – léčivo k terapii obezity – rozšíření indikací, nově: léčba obezity a kontrola hladu u dospělých a dětí od 4 let se získanou hypotalamickou obezitou (aHO) způsobenou poškozením nebo poruchou funkce hypotalamu.
mCOMBRIAX injekční disperze v předplněné injekční stříkačce, mRNA vakcína proti chřipce a onemocnění covid-19 (mRNA kódující hemaglutininové (HA) glykoproteiny sezónních chřipkových virů, mRNA kódující N-terminální doménu a receptor-vazebnou doménu spike (S) proteinu viru SARS-CoV-2) – k aktivní imunizaci pro prevenci chřipky a onemocnění covid-19 způsobeného virem SARS-CoV-2 u osob ve věku 50 let a starších. Vakcína se má používat v souladu s oficiálními doporučeními.
Mekinist 0,5 mg, 2 mg potahované tablety (trametinib) – cytostatikum, inhibitor proteinkináz – rozšíření indikací, nově: v kombinaci s dabrafenibem k léčbě dospělých pacientů s lokálně pokročilým nebo metastazujícím diferencovaným karcinomem štítné žlázy s mutací V600E genu BRAF, rezistentních nebo nezpůsobilých k léčbě radioaktivním jódem (RAI), u kterých došlo k progresi během nebo po předchozí systémové léčbě.
Noxafil 100 mg enterosolventní tablety, 300 mg koncentrát pro infuzní roztok a 300 mg enterosolventní prášek a rozpouštědlo pro perorální suspenzi (posakonazol) – antimykotikum – rozšíření indikací: nově také pro primární léčbu invazivní aspergilózy u dospělých a pediatrických pacientů od 2 let.
Olumiant 2 mg/ml perorální suspenze (baricitinib) – imunosupresivum, inhibitor Janus kináz – nová léková forma – indikace shodné s potahovanými tabletami.
Omlyclo 300 mg injekční roztok v předplněném peru (omalizumab) – nová síla ve formě předplněného pera k léčbě alergického astmatu, chronické rinosinusitidy s nosními polypy a chronické spontánní urtikarie.
Onerji 60 mg + 7,5 mg infuzní roztok (levodopa + karbidopa) – antiparkinsonikum – nová léková forma umožňující kontinuální podávání – léčba motorických fluktuací u pacientů s pokročilou Parkinsonovou chorobou (PN), které nejsou dostatečně zvládány perorálně podávanými antiparkinsoniky.
Palsonify 20 mg, 30 mg potahované tablety (paltusotin) – hormon – léčba dospělých pacientů s akromegalií.
Quadrixam 5 mg/1,25 mg/5 mg/5 mg tvrdé tobolky (perindopril-arginin, indapamid, amlodipin-besilát, bisoprolol-fumarát) – antihypertenzivum – náhrada terapie k léčbě esenciální hypertenze u dospělých pacientů, kteří již dobře odpovídají na léčbu perindoprilem, indapamidem, amlodipinem a bisoprololem podávanými současně v téže dávce, avšak jako samostatné přípravky.
Quadrixam 10 mg/2,5 mg/5 mg/5 mg tvrdé tobolky a 10 mg/2,5 mg/10 mg/5 mg (perindopril-arginin, indapamid, amlodipin-besilát, bisoprolol-fumarát) – antihypertenzivum – náhrada terapie k léčbě esenciální hypertenze u dospělých pacientů, kteří již dobře odpovídají na léčbu perindoprilem, indapamidem, amlodipinem a bisoprololem podávanými současně v téže dávce, avšak jako samostatné přípravky. Jako přídatná terapie k léčbě obtížně léčitelné hypertenze u dospělých pacientů, jejichž krevní tlak není dostatečně kontrolován perindoprilem 10 mg, indapamidem 2,5 mg a amlodipinem v maximální tolerované dávce (5 mg nebo 10 mg).
Rhapsido 25 mg potahované tablety (remibrutinib) – imunosupresivum, inhibitor BTK – léčba chronické spontánní urtikarie (CSU) u dospělých pacientů, kteří nedostatečně reagují na léčbu H1 antihistaminiky.
Sotyktu 6 mg potahované tablety (deukravacitinib) – imunosupresivum, inhibitor Janus kináz –rozšíření indikací, nově: samostatně nebo v kombinaci s methotrexátem k léčbě aktivní psoriatické artritidy (PsA) u dospělých, kteří měli nedostatečnou odpověď nebo netolerovali předchozí léčbu pomocí chorobu modifikujících antirevmatik.
Stelara 130 mg koncentrát pro infuzní roztok, 45 mg injekční roztok, 45 mg a 90 mg injekční roztok v předplněné injekční stříkačce (ustekinumab) – imunosupresivum, inhibitor interleukinu – rozšíření indikací na pediatrické pacienty od 2 let s t.hm. od 10 kg: léčba pediatrických pacientů ve věku 2 let a starších se středně těžkou až těžkou aktivní Crohnovou chorobou, u kterých buď odpověď na konvenční terapii nebo na biologickou terapii nebyla dostatečná, nebo kteří konvenční nebo biologickou léčbu netolerovali.
Tafinlar 50 mg, 75 mg tvrdé tobolky (dabrafenib) – cytostatikum, inhibitor proteinkináz – rozšíření indikací, nově: v kombinaci s trametinibem k léčbě dospělých pacientů s lokálně pokročilým nebo metastazujícím diferencovaným karcinomem štítné žlázy s mutací V600E genu BRAF, rezistentních nebo nezpůsobilých k léčbě radioaktivním jódem (RAI), u kterých došlo k progresi během nebo po předchozí systémové léčbě.
Xanirva Duo 2,5 mg/50 mg tvrdé tobolky (rivaroxaban, kyselina acetylsalicylová) – nová fixní kombinace k prevenci aterotrombotických příhod u dospělých pacientů: po akutním koronárním syndromu (AKS) se zvýšenými hladinami srdečních biomarkerů; s vysokým rizikem ischemických příhod, kteří mají ischemickou chorobu srdeční (ICHS) nebo symptomatické onemocnění periferních tepen (PAD).
Xolremdi 100 mg tvrdé tobolky (mavorixafor) – stimulans mobilizace hematopoetických kmenových buněk – u pacientů ve věku 12 let a starších k léčbě WHIM syndromu (Warts, hypogammaglobulinemia, immunodeficiency, myelokathexis syndrome; syndrom zahrnující bradavice, hypogamaglobulinemii, infekce a myelokathexii) ke zvýšení počtu cirkulujících zralých neutrofilů a lymfocytů.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
17.5.2026
Aktualizace KVĚTEN 2026
Aktualizace AISLP květen 2026 je připravena ke stažení.
Vážení uživatelé programu AISLP,
v následujícím přehledu Vám přinášíme výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální květnové verzi AISLP.
Bontens 5 mg/1,5 mg a 10 mg/1,5 mg tvrdé tobolky s řízeným uvolňováním (ramipril/indapamid) – antihypertenzivum, ACE inhibitor a diuretikum – nova fixní kombinace k léčbě esenciální hypertenze jako náhrada terapie u dospělých pacientů náležitě léčených ramiprilem a indapamidem podávanými souběžně ve stejných dávkách jako ve fixní kombinaci, ale ve formě samostatných přípravků.
Camcevi 21 mg injekční suspenze s prodlouženým uvolňováním (leuprorelin) – analog gonadorelinu – nová síla přípravku k podávání 1krát za 3 měsíce: léčba pokročilého hormonálně dependentního karcinomu prostaty a léčba vysoce rizikového, lokalizovaného a lokálně pokročilého hormonálně dependentního karcinomu prostaty v kombinaci s radioterapií.
Dupixent 200 mg a 300 mg injekční roztok v předplněné injekční stříkačce/předplněném peru (dupilumab) – dermatologikum, monoklonální protilátka – rozšíření indikací, nově: k léčbě středně těžké až těžké chronické spontánní urtikarie u dospělých, dospívajících a dětí (od 2 let) s nedostatečnou odpovědí na H1-antihistaminika a kteří dosud nebyli léčeni anti-IgE terapií CSU.
Enflonsia 105 mg injekční roztok v předplněné injekční stříkačce (klesrovimab) – antivirová monoklonální protilátka – k prevenci onemocnění dolních cest dýchacích vyvolaného respiračním syncytiálním virem (RSV) u novorozenců a kojenců během jejich první RSV sezóny. Přípravek má být používán v souladu s oficiálními doporučeními.
EURneffy 1 mg nosní sprej, roztok v jednodávkovém obalu (epinefrin) – nová síla přípravku a rozšíření indikací: k urgentní léčbě alergických reakcí (anafylaxe) způsobených bodnutím nebo štípnutím hmyzem, potravinami, léčivými přípravky a dalšími alergeny, jakož i idiopatické anafylaxe nebo anafylaxe vyvolané fyzickou námahou. Léčba je určena pro dospělé, dospívající a děti od 4 let s t.hm. 15 kg nebo vyšší.
Fylrevy 14.2 mg, 18,9 mg potahované tablety (estetrol) – gynekologikum, hormon, estrogen – hormonální substituční léčba (HST) příznaků nedostatku estrogenu u postmenopauzálních žen po hysterektomii nebo u žen s dělohou, které jsou více než 12 měsíců po menopauze.
Kygevvi 2 g/2 g prášek pro perorální roztok (doxecitin/doxribtimin) – trávicí trakt a metabolismus – léčba pediatrických a dospělých pacientů s geneticky potvrzeným deficitem thymidinkinázy 2 (TK2d) s nástupem příznaků ve věku 12 let nebo dříve.
mRESVIA injekční disperze v předplněné injekční stříkačce, mRNA vakcína (modifikovaný nukleosid) proti respiračnímu syncytiálnímu viru (RSV), zapouzdřené v lipidových nanočásticích, léčivou látkou je jednovláknová mRNA s čepičkou na 5' konci kódující glykoprotein F viru RSV-A stabilizovaný v prefuzní konformaci – rozšíření indikace: k aktivní imunizaci k prevenci onemocnění dolních cest dýchacích (lower respiratory tract disease, LRTD) způsobeného respiračním syncytiálním virem (RSV) u dospělých ve věku 18 let a starších. Vakcína se má používat v souladu s oficiálními doporučeními.
Ojemda 25 mg/ml prášek pro perorální suspenzi a 100 mg potahované tablety (tovorafenib) – cytostatikum, inhibitor proteinkináz – monoterapie pacientů od 6 měsíců s pediatrickým gliomem nízkého stupně (LGG) s fúzí nebo přestavbou genu BRAF nebo mutací BRAF V600, u kterých došlo k progresi po jedné nebo více předchozích systémových léčbách.
Penthrox 99,9% tekutina k inhalaci (methoxyfluran) – analgetikum – rozšíření indikací na pediatrické pacienty od 6 let: rychlá úleva od středně silné až silné bolesti u pacientů ve věku 6 let a starších při vědomí s traumatem a související bolestí.
Plyzari 6 mg/ml injekční roztok v předplněném peru (liraglutid) – GLP-1 agonista – rozšíření indikací, nově: u dětí (6 až < 12 let) je přípravek indikován jako doplňková léčba ke zdravé výživě a zvýšené fyzické aktivitě k úpravě tělesné hmotnosti u dětí ve věku od 6 do < 12 let s:
- obezitou (BMI >= 95. percentil) a
- tělesnou hmotností >= 45 kg.
Pokud u pacientů při dávce 3 mg/den nebo při maximální tolerované dávce nedojde po 12 týdnech k poklesu jejich BMI nebo BMI z-skóre alespoň o 4%, léčba přípravkem má být přerušena a přehodnocena.
Kalium chloratum Noridem 15% koncentrát pro infuzní roztok (chlorid draselný) – nový koncentrát k prevenci a léčbě nedostatku draslíku, pokud není možná náhrada perorálním podáním.
Kayshild 0,25 mg, 0,5 mg, 1 mg, 1,7 mg a 2,4 mg injekční roztok v předplněném peru (semaglutid) –antidiabetikum, GLP-1 agonista – ve spojení s dietou a fyzickou aktivitou k léčbě dospělých s necirhotickou steatohepatitidou asociovanou s metabolickou dysfunkcí (MASH, metabolic dysfunction-associated steatohepatitis) se středně pokročilou až pokročilou jaterní fibrózou (stadium fibrózy F2 až F3).
Kerendia 40 mg potahované tablety (finerenon) – antagonista aldosteronu – nová síla přípravku a rozšíření indikací, nově: k léčbě symptomatického chronického srdečního selhání s ejekční frakcí levé komory (LVEF) >=40% u dospělých pacientů.
Minjuvi 200 mg prášek pro koncentrát pro infuzní roztok (tafasitamab) – cytostatikum, monoklonální protilátka – rozšíření indikací, nově: v kombinaci s lenalidomidem a rituximabem k léčbě dospělých pacientů s relabujícím nebo refrakterním folikulárním lymfomem (FL) (stupeň 1-3a) po nejméně jedné linii systémové léčby.
Nuperal 10 mg/10 mg tvrdé tobolky s řízeným uvolňováním (doxylamin-hydrogen-sukcinát/pyridoxin-hydrochlorid) – nová léková forma k symptomatické léčbě nauzey a zvracení u těhotných dospělých žen, u nichž nebyla účinná konzervativní léčba.
Rezurock 200 mg potahované tablety (belumosudil) – imunosupresivum – léčba dospělých a pediatrických pacientů (od 12 let s t.hm. alespoň 40 kg) s chronickou reakcí štěpu proti hostiteli (cGVHD), u nichž jiné možnosti léčby přinášejí omezený klinický přínos, nejsou vhodné nebo byly vyčerpány.
Scemblix 100 mg potahované tablety (asciminib) – cytostatikum, inhibitor tyrozinkináz – nová síla přípravku a rozšíření indikací, nově: léčba dospělých pacientů s Ph+ CML-CP s mutací T315I, kteří jsou rezistentní, intolerantní nebo pro ně není vhodná léčba ponatinibem.
Simponi 45 mg/0,45 ml injekční roztok v předplněném peru (VarioJect) a 50 mg a 100 mg injekční roztok v předplněné injekční stříkačce (golimumab) – imunosupresivum, inhibitor tumor nekrotizujícího faktoru alfa – rozšíření pediatrických indikací, nově: k léčbě středně těžké až těžké aktivní ulcerózní kolitidy u pediatrických pacientů od 2 let s t.hm. nejméně 15 kg, u kterých odpověď na konvenční léčbu, včetně kortikosteroidů a 6-merkaptopurinu (6-MP) nebo azathioprinu (AZA), nebyla dostatečná, nebo kteří tuto léčbu netolerovali nebo byli pro tuto léčbu ze zdravotních důvodů kontraindikováni.
Supemtek injekční roztok v předplněné injekční stříkačce, trivalentní vakcína proti chřipce (rekombinantní, připravená na buněčné kultuře) – k aktivní imunizaci k prevenci chřipkového onemocnění u dospělých a dětí od 9 let. Vakcína musí být používána v souladu s oficiálními doporučeními.
Taltz 40 mg a 80 mg injekční roztok v předplněné injekční stříkačce a 80 mg v předplněném peru (ixekizumab) – imunosupresivum, inhibitor interleukinu – rozšíření pediatrických indikací, nově: juvenilní idiopatická artritida: samostatně nebo v kombinaci s methotrexátem k léčbě aktivní juvenilní psoriatické artritidy (JPsA) a aktivní artritidy související s entezitidou (ERA) u pacientů ve věku 6 let a starších a s t.hm. 25 kg, kteří nereagovali dostatečně na konvenční terapii nebo ji netolerují.
Zilbrysq 16,6 mg, 23 mg a 32,4 mg injekční roztok v předplněném peru (zilukoplan) – imunosupresivum, inhibitor komplementu – nová léková forma přípravku: přídatná léčba ke standardní léčbě generalizované myasthenia gravis (gMG) u dospělých pacientů, kteří mají pozitivní nález protilátek proti acetylcholinovým receptorům (AChR).
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
30.4.2026
Modul Duplicity rozšířen o "KiK" ve verzi pro Windows
Modul KiK je nyní k dispozici pro všechny uživatele AISLP s Windows verzí.
Modul KiK ve vašem programu AISLP
Modul "KIK", který byl zatím dostupný hlavně uživatelům velkých lékárenských systémů, je nyní dostupný pro všechny majitele licence AISLP pro Windows.
Ve spojení s již existujícím modulem Duplicity prochází zadané léčivé přípravky a výsledkem je grafické zobrazení, které Vám přehledně ukáže možné Duplicity a Kontraindikace (KiK), jak jsou uvedeny SmPC. Oba moduly nyní fungují jednoduchým vyvoláním přímo v menu, nebo je najdete pod grafickou ikonou.
Duplicity
Modul AISLP Duplicity hledá možné duplicity (i multiplicity) při výdeji léčivých přípravků, tj. zda pacient neužívá dva či více léků se stejnou nebo příbuznou léčivou látkou. Tyto duplicity nehledá jen podle ATC skupiny, ale i podle složení přípravku. Bere tak v úvahu i možnost existence více variant léčivé látky. Modul AISLP Duplicity spolehlivě najde léčivou látku ve všech přípravcích, ve kterých se vyskytuje, tedy i v kombinovaných přípravcích a různých lékových formách.
KiK
KiK slouží jako rychlé upozornění uživatele na možné kontraindikované kombinace léčivých přípravků, které jsou mu předány Vaším lékárenským/ambulantním softwarem formou SÚKL kódů. Modul vychází z informací uvedených v SmPC léčivých přípravků a rychle prověří možné kontraindikace zadaných léčivých přípravků, ať už přímo při výdeji v lékárně, předepisování v ordinaci lékaře nebo při výběru léčivých přípravků z historie pacienta. KiK doplňuje AISLP a umožňuje zobrazit grafický výstup přímo z Vašeho softwaru.
Více o možnostech modulů Duplicity a KiK včetně příkladů naleznete v naší Studovně.
Váš tým AISLP
www.aislp.cz
16.4.2026
Aktualizace DUBEN 2026
Aktualizace AISLP duben 2026 je připravena ke stažení.
Vážení uživatelé programu AISLP,
přinášíme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální dubnové verzi AISLP.
Anktiva 400 mikrogramů koncentrát pro intravezikální suspenzi (nogapendekin alfa a inbakicept ) – imunostimulans, interleukin – v kombinaci s Bacillus Calmette-Guérin (BCG) k léčbě dospělých pacientů s neinvazivním karcinomem močového měchýře, který neinfiltruje svalovinu močového měchýře (NMIBC; non-muscle invasive bladder cancer), s karcinomem in situ (CIS), s papilárními nádory nebo bez nich, který nereaguje na BCG.
Arexvy prášek a suspenze pro injekční suspenzi, vakcína proti respiračnímu syncytiálnímu viru (RSV), rekombinantní, obsahující adjuvans (RSVPreF3 antigen, rekombinantní glykoprotein F respiračního syncytiálního viru stabilizovaný v prefuzní konformaci) – rozšíření indikace: k aktivní imunizaci za účelem prevence onemocnění dolních cest dýchacích způsobeného respiračním syncytiálním virem u dospělých ve věku 18 let a starších. Použití vakcíny má být v souladu oficiálními doporučeními.
Iclusig 15 mg, 30 mg a 45 mg potahované tablety (ponatinib) – cytostatikum, inhibitor tyrozinkináz – rozšíření indikací, nově: léčba dospělých pacientů s nově diagnostikovanou Ph+ ALL v kombinaci s chemoterapií se sníženou intenzitou.
Imfinzi 50 mg/ml koncentrát pro infuzní roztok (durvalumab) – cytostatikum, monoklonální protilátka – rozšíření indikací, nově: v kombinaci s chemoterapií na bázi platiny jako neoadjuvantní léčba následovaná monoterapií jako adjuvantní léčba k léčbě dospělých pacientů s resekovatelným nemalobuněčným karcinomem plic (NSCLC) s vysokým rizikem recidivy a bez mutací EGFR nebo přestavby ALK; monoterapie dospělých pacientů s malobuněčným karcinomem plic v limitovaném stadiu (LS-SCLC), u kterých nedošlo k progresi onemocnění po chemoradiační léčbě na bázi platiny; v kombinaci s gemcitabinem a cisplatinou jako neoadjuvantní léčba, následovaná monoterapií jako adjuvantní léčba po radikální cystektomii k léčbě dospělých pacientů s resekovatelným invazivním karcinomem močového měchýře (MIBC).
Mounjaro 2,5 mg, 5 mg, 7,5 mg, 10 mg, 12,5 mg a15 mg injekční roztok v předplněném peru – (tirzepatid) – antidibetikum, duální GIP a GLP-1 agonista – rozšíření indikací pro pediatrickou populaci k léčbě DM 2. typu: k léčbě dospělých, dospívajících a dětí ve věku 10 let a starších s nedostatečně kontrolovaným diabetes mellitus 2. typu jako doplněk k dietě a cvičení: v monoterapii, pokud není podávání metforminu vhodné z důvodu nesnášenlivosti nebo kontraindikací; jako přídavná léčba k dalším léčivým přípravkům k léčbě diabetu.
Recarbrio 500 mg/500 mg/250 prášek pro infuzní roztok (imipenem/cilastatin/relebaktam) – beta-laktamové antibiotikum ze skupiny karbapenemů – rozšíření indikací: k léčbě dospělých a pediatrických pacientů od narození s nozokomiální pneumonií (HAP), včetně ventilátorové pneumonie (VAP); k léčbě bakteriemie, která se vyskytuje ve spojení s HAP nebo VAP nebo, u které existuje podezření na tuto souvislost; k léčbě infekcí vyvolaných aerobními gramnegativními mikroorganismy s omezenými terapeutickými možnostmi. Je nutno dbát doporučených postupů pro správné používání antibakteriálních látek.
Sacubitril/Valsartan Polpharma 24 mg/26 mg, 49 mg/51 mg a 97 mg/103 mg potahované tablety – léčba symptomatického chronického srdečního selhání se sníženou ejekční frakcí u dospělých pacientů.
Valkubit 24 mg/26 mg, 49 mg/51 mg a 97 mg/103 mg potahované tablety (sakubitril/valsartan) – léčba symptomatického chronického srdečního selhání se sníženou ejekční frakcí u dospělých pacientů. Léčba symptomatického chronického srdečního selhání se systolickou dysfunkcí levé komory u dětí od 1 roku věku a dospívajících.
Wegovy 0,25 mg, 0,5 mg, 1 mg, 1,7 mg a 2,4 mg injekční roztok v předplněném peru (semaglutid) – GLP-1 agonista – aktualizace SmPC a PIL týkající se úpravy tělesné hmotnosti u dospělých pacientů pomocí vyšší dávky semaglutidu (7,2 mg jednou týdně po minimálně 4 týdnech podávání dávky 2,4 mg jednou týdně u dospělých s BMI >= 30 kg/m2 na začátku léčby).
Zynyz 500 mg koncentrát pro infuzní roztok (retifanlimab) – cytostatikum, monoklonální protilátka – rozšíření indikací, nově: v kombinaci s karboplatinou a paklitaxelem k léčbě první linie u dospělých pacientů s metastazujícím nebo neoperabilním lokálně rekurentním dlaždicobuněčným karcinomem análního kanálu (SCAC).
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
12.3.2026
Aktualizace BŘEZEN 2026
Aktualizace AISLP březen 2026 je připravena ke stažení.
Vážení uživatelé programu AISLP,
v následujícím přehledu Vám přinášíme výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální březnové verzi AISLP.
Aqneursa 1 g granule pro perorální suspenzi v sáčku (levacetylleucin) – léčivo nervového systému – léčba neurologických projevů Niemannovy-Pickovy choroby typu C (NPC) v kombinaci s miglustatem nebo jako monoterapie u pacientů, kteří miglustat netolerují, u dospělých a dětí ve věku od 6 let s t.hm. nejméně 20 kg.
Aumseqa 55 mg potahované tablety (aumolertinib-mesilát) – cytostatikum, inhibitor proteinkináz – v monoterapii k léčbě v první linii u dospělých pacientů s pokročilým nemalobuněčným karcinomem plic (NSCLC), jejichž nádory mají delece exonu 19 genu EGFR nebo substituční mutaci exonu 21 (L858R) genu EGFR; k léčbě dospělých pacientů s pokročilým NSCLC s pozitivní mutací T790M genu EGFR.
Axhidrox 8 mg/g krém (glykopyrronium-bromid) – dermatologikum, antihidrotikum – rozšíření indikace: k lokální léčbě těžké primární axilární hyperhidrózy u dospělých a dospívajících od 12 let.
Dovprela 200 mg tablety (pretomanid) – antituberkulotikum – aktualizace SmPC, změna indikací: v kombinaci s bedachilinem, linezolidem a moxifloxacinem k léčbě dospělých s plicní tuberkulózou (TBC) způsobenou Mycobacterium tuberculosis rezistentní na rifampicin, s rezistencí na isoniazid nebo bez ní. V kombinaci s bedachilinem a linezolidem k léčbě dospělých s plicní TBC způsobenou M. tuberculosis rezistentní na rifampicin a fluorochinolon, s rezistencí na isoniazid nebo bez ní.
Exdensur 100 mg injekční roztok v předplněném peru
Exdensur 100 mg injekční roztok v předplněné injekční stříkačce (depemokimab) – antiastmatikum –doplňková udržovací léčba těžkého astmatu se zánětem typu 2 charakterizovaným počtem eozinofilů v krvi u dospělých a dospívajících od 12 let, kteří nedosahují dostatečné kontroly astmatu navzdory vysoké dávce inhalačních kortikosteroidů v kombinaci s dalším léčivým přípravkem na kontrolu astmatu; doplňková léčba k intranazálním kortikosteroidům k léčbě dospělých pacientů s těžkou chronickou rinosinusitidou s nosními polypy, u nichž léčba systémovými kortikosteroidy a/nebo chirurgický zákrok nevedou k dosažení dostatečné kontroly onemocnění.
Gazyvaro 1000 mg koncentrát pro infuzní roztok (obinutuzumab) – cytostatikum, monoklonální protilátka – rozšíření indikací, nově: v kombinaci s mofetil-mykofenolátem (MMF) k léčbě dospělých pacientů s aktivní lupusovou nefritidou (LN) třídy III nebo IV, se souběžnou třídou V či bez ní.
Loteprednol Olikla 5 mg/ml oční kapky, suspenze (loteprednol-etabonát) – oftalmologikum, kortikosteroid – léčba pooperačního zánětu po oční operaci.
mNEXSPIKE injekční disperze v předplněné injekční stříkačce, mRNA vakcína proti onemocnění covid-19 (mRNA kódující N-terminální doménu a receptor-vazebnou doménu spike (S) proteinu viru SARS-CoV-2 (XBB.1.5)) – k aktivní imunizaci osob ve věku 12 let a starších k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2. Vakcína se má používat v souladu s oficiálními doporučeními.
Myqorzo 5 mg, 10 mg, 15 mg a 20 mg potahované tablety (afikamten) – kardiakum – k léčbě symptomatické (New York Heart Association, NYHA, třída II-III) hypertrofické obstrukční kardiomyopatie (oHCM) u dospělých pacientů.
Orebriton Duo 90 mg/50 mg tvrdé tobolky (tikagrelor, kyselina acetylsalidylová) – protidestičkové léčivo – nová fixní kombinace k prevenci aterotrombotických příhod u dospělých pacientů s akutním koronárním syndromem (ACS).
Pitezenib 2 mg/10 mg a 4 mg/10 mg tablety (pitavastatin, ezetimib) – hypolipidemikum – náhrada terapie doplňující dietu a další nefarmakologickou léčbu u dospělých pacientů s primární hypercholesterolemií, včetně heterozygotní familiární a nefamiliární hypercholesterolemie nebo kombinované (smíšené) dyslipidemie, kteří jsou dostatečně kompenzováni pitavastatinem a ezetimibem podávanými současně ve stejné dávce jako ve fixní kombinaci dávek, ale jako samostatné přípravky.
Salbutamol Noridem 2,5 mg/2,5 ml roztok k rozprašování
Salbutamol Noridem 5 mg/2,5 ml roztok k rozprašování – léčba akutního těžkého astmatu a léčba těžkých akutních exacerbací chronické obstrukční plicní nemoci (CHOPN) u dospělých, dospívajících a dětí od 4 let.
Salbutamol Noridem 1,25 mg/2,5 ml roztok k rozprašování – léčba akutního těžkého astmatu u dětí a kojenců.
Uplizna 100 mg koncentrát pro infuzní roztok (inebilizumab) – imunosupresivum, monoklonální protilátka – nová indikace: doplněk standardní léčby generalizované myasthenia gravis (gMG) u dospělých pacientů, kteří jsou pozitivní na protilátky proti acetylcholinovému receptoru (AChR) nebo proti svalově specifické tyrozin-kináze (MuSK).
Vueway 0,5 mmol/ml injekční roztok (gadopiklenol) – diagnostikum, paramagnetická kontrastní látka – rozšíření indikací na pediatrické pacienty od narození.
Xerava 50 mg a 100 mg prášek pro koncentrát pro infuzní roztok (eravacyklin) – tetracyklinové antibiotikum – rozšíření indikace: k léčbě komplikovaných intraabdominálních infekcí (cIAI) u dospělých a dospívajících od 12 let s t.hm. nejméně 50 kg.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
12.2.2026
Aktualizace ÚNOR 2026
Aktualizace AISLP únor 2026 je připravena ke stažení.
Vážení uživatelé programu AISLP,
přinášíme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální únorové verzi AISLP.
Zároveň v aktuální verzi AISLP naleznete v našem seznamu nové ATC skupiny a jejich změny platné pro rok 2026, jak je uvádí WHO Collaborating Centre for Drug Statistics Methodology. Zařazení léčivých látek a léčivých přípravků je již v souladu s ATC/DDD Indexem 2026.
Austedo 12 mg, 24 mg, 30 mg, 36 mg, 42 mg a 48 mg tablety s prodlouženým uvolňováním (deutetrabenazin) – léčivo nervového systému – léčba středně těžké až těžké tardivní dyskineze u dospělých.
Cejemly 600 mg koncentrát pro infuzní roztok (sugemalimab) – cytostatikum, monoklonální protilátka – rozšíření indikací, nově: v monoterapii k léčbě neresekabilního NSCLC stadia III bez senzibilizujících mutací EGFR nebo genomových aberací ALK, ROS1 u dospělých, jejichž nádory exprimují PD-L1 na ≥1% nádorových buněk a u jejichž onemocnění nedošlo k progresi po chemoradioterapii na bázi platiny.
Cidofovir Tillomed 75 mg/ml koncentrát pro infuzní roztok (dihydrát cidofoviru) – antivirotikum – k léčbě CMV retinitidy u dospělých se syndromem získané imunodeficience (AIDS) a bez poruchy renální funkce. Cidofovir se má používat pouze v případech, kdy jsou jiné léky považovány za nevhodné.
Clozapine Desitin 25 mg a 100 mg (klozapin) – antipsychotikum – upravená doporučení pro rutinní sledování krevního obrazu, aktualizace SmPC.
Dawnzera 80 mg injekční roztok v předplněném peru (donidalorsen) – antisense oligonukleotid k rutinní prevenci rekurentních atak hereditárního angioedému (HAE) u dospělých a dospívajících od 12 let.
Dupixent 200 mg a 300 mg injekční roztok v předplněné injekční stříkačce/předplněném peru (dupilumab) – dermatologikum, monoklonální protilátka – rozšíření indikací, nově: k léčbě středně těžké až těžké chronické spontánní urtikarie u dospělých a dospívajících (od 12 let) pacientů, kteří mají nedostatečnou odpověď na H1-antihistaminika a kteří dosud nebyli léčeni anti-IgE terapií CSU.
Inluriyo 200 mg potahované tablety (imlunestrant) – cytostatikum, antiestrogen – monoterapie dospělých pacientů s lokálně pokročilým nebo metastazujícím karcinomem prsu pozitivním na estrogenové receptory (ER), HER2-negativním, s aktivační mutací ESR1, kteří mají progresi onemocnění po předchozím režimu s endokrinní léčbou.
Koselugo 5 mg a 7,5 mg granule v tobolce k otevření (selumetinib) – cytostatikum, inhibitor proteinkináz – nová léková forma přípravku: v monoterapii k léčbě symptomatických, neoperovatelných plexiformních neurofibromů (PN) u pacientů s neurofibromatózou 1. typu (NF1) od 1 roku do 7 let a starších pacientů, kteří mají obtíže s polykáním.
Leponex 25 mg, 100 mg a 200 mg tablety (klozapin) – antipsychotikum – upravená doporučení pro rutinní sledování krevního obrazu, aktualizace SmPC.
Shingrix injekční suspenze v předplněné injekční stříkačce, vakcína proti herpes zoster (rekombinantní, adjuvovaná), nová léková forma – prevence herpes zoster (HZ) a postherpetické neuralgie (PHN) u dospělých ve věku 50 let a starších; u dospělých od 18 let se zvýšeným rizikem HZ. Použití vakcíny se má řídit místním oficiálním doporučením.
Teizeild 1 mg/ml koncentrát pro infuzní roztok (teplizumab) – antidiabetikum, monoklonální protilátka – k oddálení nástupu 3. stadia diabetu 1. typu (diabetes mellitus 1. typu - DM1T) u dospělých a pediatrických pacientů ve věku od 8 let s DM1T ve 2. stadiu.
Tremfya 45 mg/0,45 ml injekční roztok v předplněném peru (guselkumab) – imunosupresivum, inhibitor interleukinu – nová síla a léková forma vhodná pro pediatrické pacienty: léčba středně těžké až těžké plakové psoriázy u dětí a dospívajících od 6 let věku, kteří jsou kandidáty pro systémovou léčbu.
Uplizna 100 mg koncentrát pro infuzní roztok (inebilizumab) – imunosupresivum, monoklonální protilátka – nová indikace: léčba dospělých pacientů s aktivní imunoglobulin G4 asociovanou nemocí (IgG4-RD).
VacPertagen injekční suspenze v předplněné injekční stříkačce, vakcína proti černému kašli (rekombinantní, acelulární komponenta, adsorbovaná) – k posílení imunizace proti pertusi u osob ve věku 12 let a starších; k pasivní ochraně proti pertusi v raném dětství po imunizaci matky během těhotenství. Vakcína se má používat v souladu s oficiálními doporučeními.
Veyvondi 650 IU a 1300 IU prášek a rozpouštědlo pro injekční roztok (vonikog alfa) – rozšíření indikace pro děti mladší 18 let s von Willebrandovou chorobou k léčbě krvácení, pokud je léčba desmopresinem neúčinná nebo je kontraindikována.
Waskyra 2-10 milionů buněk/ml infuzní disperze (etuvetidigen autotemcel) – přípravek genové terapie k léčbě pacientů ve věku od 6 měsíců s Wiskottovým-Aldrichovým syndromem (WAS), kteří mají mutaci v genu WAS, u kterých je vhodná transplantace hematopoetických kmenových buněk (HSCT) a není k dispozici příbuzný dárce HSC s odpovídajícím HLA.
Wayrilz 400 mg potahované tablety (rilzabrutinib) – inhibitor Brutonovy tyrozinkinázy – léčba imunitní trombocytopenie (ITP) u dospělých pacientů, kteří jsou refrakterní na jinou léčbu.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
20.1.2026
Leden s AISLP
Lednová akce na licence AISLP.
Pořiďte si licenci AISLP a od nás dostanete hned dva praktické dárky.
- mobilní verzi AISLP na rok zdarma
- stylový fitness ručník, ideální pro plnění Vašich novoročních sportovních předsevzetí :).
Stačí vyplnit registrační formulář.
Přejeme Vám krásný začátek roku 2026.
Váš tým AISLP
www.aislp.cz

15.1.2026
Aktualizace LEDEN 2026
Aktualizace AISLP leden 2026 je připravena ke stažení.
Vážení uživatelé programu AISLP,
přejeme Vám, za celý náš tým, úspěšný vstup do roku 2026 a přinášíme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální lednové verzi AISLP.
Digoxin ENEO 0,05 mg/ml perorální roztok (digoxin) – kardiotonikum – k léčbě srdečního selhání, supraventrikulárních poruch rytmu: zpomalení nebo snížení fibrilace síní nebo flutteru síní. Přípravek je indikován k léčbě dětí.
Etofenamát Greencango 100 mg/g gel (etofenamát) – nesteroidní antiflogistikum k lokální aplikaci – lokální symptomatická léčba mírné až středně silné bolesti při akutních podvrtnutích, nataženích a zhmožděninách v oblasti končetin po tupém traumatu (např. sportovních úrazech) nebo mírné až středně silné bolesti měkkých tkání v blízkosti kloubu (např. burzy, šlachy, vazy a kloubní pouzdra) při osteoartróze kolenních kloubů. Pro dospělé.
Imaavy 85 mg/ml koncentrát pro infuzní roztok (nipokalimab) – imunosupresivum, inhibitor neonatálního Fc receptoru – přídatná terapie generalizované myasthenie gravis (gMG) u dospělých a dospívajících pacientů ve věku 12 let a starších, kteří mají pozitivní nález protilátek proti acetylcholinovému receptoru (AChR) nebo proti svalově specifické tyrosinkináze (MuSK).
Invokana 100 mg a 300 mg potahované tablety (hemihydrát kanagliflozinu) – antidiabetikum, inhibitor SGLT2 – rozšíření indikací o pediatrickou populaci: k léčbě dospělých a dětí ve věku 10 let a starších s nedostatečně kontrolovaným diabetes mellitus 2. typu jako přídatná terapie k dietě a cvičení: v monoterapii, pokud se metformin považuje za nevhodný v důsledku nesnášenlivosti nebo kontraindikací; jako přídatná léčba k dalším léčivým přípravkům k léčbě diabetu.
Kyinsu (700 jednotek + 2 mg)/ml injekční roztok v předplněném peru (insulin-ikodek, semaglutid) – antidiabetikum – k léčbě dospělých s diabetes mellitus 2. typu nedostatečně kontrolovaných bazálním inzulinem nebo agonisty receptoru glukagonu podobného peptidu 1 (GLP-1) jako doplněk diety a cvičení v kombinaci s perorálními antidiabetiky.
Libtayo 350 mg koncentrát pro infuzní roztok (cemiplimab) – cytostatikum, monoklonální protilátka – rozšíření indikací: nově v monoterapii k adjuvantní léčbě dospělých pacientů se spinocelulárním karcinomem kůže s vysokým rizikem recidivy po operaci a ozařování.
Lynkuet 60 mg měkké tobolky (elinzanetant) – gynekologikum – léčba středně těžkých až těžkých vazomotorických příznaků (VMS): spojených s menopauzou; způsobených adjuvantní endokrinní terapií (AET) související s karcinomem prsu.
Paxlovid 150 mg + 100 mg potahované tablety (nirmatrelvir + ritonavir) – antivirotikum – rozšíření indikace o pediatrickou populaci: k léčbě onemocnění covid-19 u dospělých a pediatrických pacientů ve věku 6 let a starších s t.hm. nejméně 20 kg, kteří nevyžadují doplňkovou léčbu kyslíkem a u kterých je zvýšené riziko progrese do závažné formy onemocnění covid-19.
Prolastina 1000 mg, 4000 mg a 5000 mg prášek a rozpouštědlo pro infuzní roztok (lidský inhibitor alfa1-proteinasy) – nová síla (koncentrace po rekonstituci je 25 mg/ml) k dlouhodobé augmentační léčbě osob s prokázaným těžkým deficitem inhibitoru alfa1-proteázy.
Remsima 40 mg/ml koncentrát pro infuzní roztok (infliximab) – imunosupresivum, inhibitor tumor nekrotizujícího faktoru alfa – nová léková forma přípravku – revmatoidní artritida u dospělých. Crohnova choroba u dospělých a pediatrických pacientů od 6 let. Ulcerózní kolitida u dospělých a pediatrických pacientů od 6 let. Ankylozující spondylitida u dospělých. Psoriatická artritida u dospělých. Psoriáza u dospělých.
Rivaroxaban Koanaa 10 mg, 15 mg a 20 mg filmy dispergovatelné v ústech (rivaroxaban) – nová léková forma bioekvivalentní s potahovanými tabletami.
Saphnelo 120 mg injekční roztok v předplněné injekční stříkačce/peru (anifrolumab) – imunosupresivum, monoklonální protilátka – nová léková forma přípravku – přídatná terapie k léčbě dospělých pacientů se středně těžkým až těžkým aktivním systémovým lupus erythematodes (SLE) s pozitivitou autoprotilátek i přes standardní terapii.
Sirturo 20 mg a 100 mg tablety (bedachilin-fumarát) – antituberkulotikum – rozšíření indikace: jako součást vhodné kombinované léčby u dospělých a pediatrických pacientů (od 2 let s t.hm. nejméně 7 kg) s plicní tuberkulózou (TB) vyvolanou Mycobacterium tuberculosis rezistentní přinejmenším na rifampicin a isoniazid. Je nutné respektovat oficiální pokyny pro správné používání antituberkulotik.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
15.12.2025
Aktualizace PROSINEC 2025
Aktualizace AISLP prosinec 2025 je připravena ke stažení.
Vážení uživatelé programu AISLP,
přejeme Vám příjemně strávený adventní čas a přinášíme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální prosincové verzi AISLP.
Amfotericin B liposomal Tillomed 50 mg prášek pro koncentrát pro infuzní disperzi (amfotericin B v lipozomální formě) – antimykotikum – přípravek je indikován u dospělých a pediatrických pacientů od 1 měsíce věku: k léčbě závažných systémových nebo hlubokých mykóz; k empirické léčbě suspektních mykotických infekcí u pacientů s febrilní neutropenií. Přípravek lze použít jako sekundární léčbu viscerální leishmaniózy (Leishmania donovani) u imunokompetentních pacientů a u pacientů s oslabeným imunitním systémem (např. u osob infikovaných HIV). U pacientů s oslabeným imunitním systémem je nutné počítat s rekurencí. Nejsou dostupné žádné údaje o prevenci rekurence. Je nutné zohlednit národní a mezinárodní doporučení týkající se vhodného používání antimykotik.
Brinsupri 25 mg potahované tablety (brensokatib) – inhibitor dipeptidylpeptidázy 1 (DPP1) – léčba bronchiektázie bez přítomnosti cystické fibrózy u pacientů od 12 let se dvěma nebo více exacerbacemi v předchozích 12 měsících.
Calquence 100 mg tvrdé tobolky (akalabrutinib) – cytostatikum, inhibitor proteinkináz – rozšíření indikací, nově: v kombinaci s venetoklaxem s obinutuzumabem nebo bez obinutuzumabu k léčbě dospělých pacientů s dosud neléčenou chronickou lymfocytární leukemií (CLL); v kombinaci s bendamustinem a rituximabem (BR) k léčbě dospělých pacientů s dosud neléčeným lymfomem z plášťových buněk (MCL), kteří nejsou vhodní pro autologní transplantaci krvetvorných kmenových buněk (ASCT); v monoterapii k léčbě dospělých pacientů s relabujícím nebo refrakterním lymfomem z plášťových buněk (MCL), kteří dříve nebyli léčeni inhibitorem BTK.
Humira 40 mg/0,8 ml injekční roztok (adalimumab) – imunosupresivum, inhibitor tumor nekrotizujícího faktoru – zrušení lékové formy (injekční lahvička), pro pediatrické pacienty je dále určena Humira 20 mg injekční roztok v předplněné injekční stříkačce.
Keytruda 25 mg/ml koncentrát pro infuzní roztok (pembrolizumab) – cytostatikum, monoklonální protilátka – rozšíření indikací, nově: v monoterapii k neoadjuvantní léčbě resekovatelného lokálně pokročilého skvamózního karcinomu hlavy a krku s následnou adjuvantní léčbou v kombinaci s radioterapií a se současným podáváním cisplatiny nebo bez ní a poté v monoterapii u dospělých, jejichž nádory exprimují PD-L1 s CPS 1 a více.
Keytruda 395 mg a 790 mg injekční roztok – nová léková forma určená pro dospělé pacienty, shodné indikace jako u koncentrátu pro infuzní roztok.
Omlyclo 300 mg injekční roztok v předplněné injekční stříkačce (omalizumab) – nová síla přípravku: alergické astma (vyvolané IgE) u dospělých a dospívajících (od 12 let): doplňková léčba ke zlepšení kontroly astmatu u pacientů s těžkým perzistujícím alergickým astmatem, kteří mají pozitivní kožní test nebo reaktivitu in vitro na celoroční vzdušný alergen a kteří mají sníženou funkci plic (FEV1 <80%), stejně jako časté symptomy během dne nebo noční probouzení, a kteří mají prokázané četné těžké exacerbace astmatu navzdory vysokým denním dávkám inhalačních kortikosteroidů a dlouhodobě působících inhalačních beta2-agonistů. Chronická rinosinusitida s nosními polypy: doplňková léčba s intranazálními kortikosteroidy k léčbě dospělých (od 18 let) se závažnou chronickou rinosinusitidou s nosními polypy, u kterých léčba intranazálními kortikosteroidy neposkytuje odpovídající kontrolu onemocnění. Chronická spontánní urtikarie (CSU): doplňková léčba k léčbě chronické spontánní urtikarie u dospělých a dospívajících pacientů (12 let a více) s nedostatečnou odpovědí na léčbu H1-antihistaminiky.
Pedismof Newborn infuzní emulze – parenterální výživa pro novorozence (i předčasně narozené)
Pedismof Infant infuzní emulze – parenterální výživa pro novorozence a malé děti do 2 let
Pedismof Pediatric infuzní emulze – parenterální výživa pro novorozence, kojence, batolata, děti a dospívající. Tříkomorový vakový systém umožňuje smísení pouze dvou komor (aminokyseliny s/bez elektrolytů + glukóza), pokud je podávání lipidů nežádoucí.
Winlevi 10 mg/g krém (klaskoteron) – dermatologikum, inhibitor androgenních receptorů – dospělí: k léčbě acne vulgaris; dospívající (od 12 let do <18 let): k léčbě acne vulgaris na obličeji.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
9.12.2025
Vánoční balíček AISLP
Vánoční akce na licence AISLP.
Pořiďte si vánoční balíček AISLP a od nás dostanete hned dva praktické dárky.
- mobilní verzi aplikace na celý rok zdarma
- stylový fitness ručník, ideální pro plnění Vašich novoročních sportovních předsevzetí :).
Stačí vyplnit registrační formulář.
Přejeme Vám krásný adventní čas.
Váš tým AISLP
www.aislp.cz
1.12.2025
Moduly AISLP Duplicity a KiK v systému Mediox
Moduly AISLP Duplicity a KiK jsou dostupné v lékárenském systému Mediox.
AISLP je informační systém (databáze) léčivých přípravků registrovaných v ČR, schvalovaných SÚKL i centralizovanou procedurou (EMA), parafarmaceutik a prostředků zdravotnické techniky. Je možné ji propojit s dalšími ambulantními, nemocničními a lékárenskými systémy.
Jedním z takových lékárenských systémů je i Mediox, jejímž výrobcem je firma Apatyka servis s.r.o.. V systému Mediox jsou nyní dostupné moduly AISLP Duplicity a KiK.
Více o propojení AISLP a Mediox si můžete přečíst přímo na stránkách
Apatyka Servis.
O možnostech modulů Duplicity a KiK se více dozvíte v naší
Studovně.
Stále platí nabídka pro nové uživatele programu AISLP - vyzkoušejte si KIK a Duplicity za výhodných podmínek.
Pokud si objednáte licenci AISLP do konce roku 2025, zaplatíte za každý měsíc do konce roku 2025 jen 1 Kč. Objednejte co nejdříve, ať plně využijete tuto speciální akci.
Tato akce se týká nových uživatelů programu AISLP. Akceptací služby vzniká závazek na 24 měsíců. Akce trvá do 31.12.2025 včetně.
Pro účast na této akci je nutno se registrovat.
V případě zájmu nás neváhejte kontaktovat na tel. 777 746 490 nebo na emailu: info@aislp.cz.
Váš tým AISLP
www.aislp.cz

12.11.2025
Aktualizace LISTOPAD 2025
Aktualizace AISLP listopad 2025 je připravena ke stažení.
Vážení uživatelé programu AISLP,
v následujícím přehledu Vám přinášíme výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální listopadové verzi AISLP.
Abrysvo prášek a rozpouštědlo pro injekční roztok (stabilizovaný prefuzní F antigen RSV podskupiny A a stabilizovaný prefuzní F antigen RSV podskupiny B), vakcína proti respiračnímu syncytiálnímu viru (bivalentní, rekombinantní) – aktualizace bodu 4.2, 4.8 a 5.1 SmPC týkající se očkování u imunokompromitovaných osob (od 18 let věku).
Benlysta 200 mg injekční roztok v předplněném peru (belimumab) – imunosupresivum, monoklonální protilátka, rozšíření indikace na pediatrické pacienty od 5 let s t.hm. od 15 kg – přídatná léčba u pacientů ve věku od 5 let s aktivním systémovým lupus erythematodes (SLE) s pozitivními autoprotilátkami a vysokým stupněm aktivity onemocnění (např. pozitivní protilátky proti dvouvláknové DNA (anti-dsDNA) a nízká hladina komplementu) navzdory standardní léčbě.
Bimervax injekční emulze (selvakovatein)
Bimervax XBB.1.16 injekční emulze (damlekovatein)
vakcína proti onemocnění covid-19 (rekombinantní, obsahující adjuvans) – rozšíření indikací, nově: k aktivní imunizaci k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2 u osob ve věku 12 let a starších. Použití vakcíny má být v souladu s oficiálními doporučeními.
Bimervax LP.8.1 injekční emulze (fúzní homodimer rekombinantní receptorové vazebné domény spike proteinu viru SARS-CoV-2 (kmen Omikron LP.8.1 - LP.8.1) navázaný na adjuvans SQBA), vakcína proti onemocnění covid-19 (rekombinantní, obsahující adjuvans) – k aktivní imunizaci k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2 u osob ve věku 12 let a starších. Použití vakcíny má být v souladu s oficiálními doporučeními.
Cabometyx 20 mg, 40 mg, 60 mg potahované tablety (kabozantinib) – cytostatikum, inhibitor proteinkináz – rozšíření indikací, nově: léčba dospělých pacientů s neresekovatelnými nebo metastazujícími, dobře diferencovanými extrapankreatickými (epNET) a pankreatickými (pNET) neuroendokrinními nádory, u kterých došlo k progresi onemocnění po alespoň jedné předchozí systémové léčbě jiné než léčbě analogy somatostatinu.
Columvi 2,5 mg, 10 mg koncentrát pro infuzní roztok (glofitamab) – cytostatikum, monoklonální protilátka – rozšíření indikací, nově: v kombinaci s gemcitabinem a oxaliplatinou k léčbě dospělých pacientů s blíže neurčeným relabujícím nebo refrakterním difuzním velkobuněčným B-lymfomem (DLBCL NOS), kteří nejsou vhodní k autologní transplantaci krvetvorných buněk (ASCT).
Ekterly 300 mg potahované tablety (sebetralstat) – symptomatická léčba akutních atak hereditárního angioedému (HAE) u dospělých a dospívajících od 12 let.
Ilaris 150 mg injekční roztok v předplněném peru (kanakinumab) – imunosupresivum, inhibitor interleukinu – nová léková forma přípravku.
Imreplys 250 mikrogramů prášek pro injekční roztok (sargramostim) – hemopoetický růstový faktor – léčba pacientů všech věkových kategorií akutně vystavených myelosupresivním dávkám záření s hematopoetickým subsyndromem akutního radiačního syndromu (H-ARS, Haematopoietic Sub-syndrome of Acute Radiation Syndrome). Přípravek má být používán v souladu s oficiálními radiologickými/nukleárními doporučeními pro mimořádné situace.
Kisunla 350 mg koncentrát pro infuzní roztok (donanemab) – biologická léčba demence – léčba dospělých pacientů s klinickou diagnózou mírné kognitivní poruchy a mírné demence v důsledku Alzheimerovy choroby (časná symptomatická Alzheimerova choroba), kteří jsou heterozygoty nebo nejsou nositeli genu pro apolipoprotein E epsilon 4 (ApoE4) s potvrzenou amyloidovou patologií.
Lymira 0,5 mg kit pro radiofarmakum (lidský albumin ve formě nanokoloidu) – radionuklidové diagnostikum – po značení roztokem technecistanu-(99mTc) sodného je získaný roztok indikován u dospělých a u pediatrické populace pro:
intravenózní podání: zobrazení kostní dřeně (přípravek není vhodný k vyšetření hematopoetické aktivity kostní dřeně) a zobrazení zánětů v jiných oblastech než břicho;
subkutánní podání: lymfoscintigrafie k prokázání neporušenosti lymfatického systému a odlišení obstrukce žilního a lymfatického systému; k předoperačnímu zobrazování a intraoperační detekci sentinelových lymfatických uzlin u melanomu, karcinomu prsu, karcinomu penisu, skvamózního karcinomu dutiny ústní a karcinomu vulvy.
Romvimza 14 mg, 20 mg, 30 mg tvrdé tobolky (vimseltinib) – cytostatikum, inhibitor proteinkináz – léčba dospělých pacientů se symptomatickým tenosynoviálním obrovskobuněčným nádorem (TGCT) souvisejícím s klinicky významným zhoršením tělesné funkce, u nichž byly možnosti chirurgického řešení vyčerpány nebo by vedly k nepřijatelné morbiditě nebo invaliditě.
Spikevax LP.8.1 25 mikrogramů injekční disperze v předplněné injekční stříkačce
mRNA vakcína proti onemocnění covid-19 (mRNA kódující kódující spike protein viru SARS-CoV-2 - LP.8.1) – k aktivní imunizaci jedinců od 6 měsíců věku k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2. Vakcínu je třeba používat v souladu s oficiálními doporučeními.
Tezspire 210 mg injekční roztok v předplněné injekční stříkačce
Tezspire 210 mg injekční roztok v předplněném peru (tezepelumab) – rozšíření indikace: přídavná léčba s intranazálními kortikosteroidy k léčbě dospělých pacientů s těžkou chronickou rinosinusitidou s nosními polypy, u kterých léčba systémovými kortikosteroidy a/nebo chirurgický zákrok neposkytují dostatečnou kontrolu onemocnění.
Zurzuvae 20 mg, 35 mg a 30 mg tvrdé tobolky (zuranolon) – antidepresivum – léčba poporodní deprese (postpartum depression, PPD) u dospělých žen po porodu.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
15.10.2025
Aktualizace ŘÍJEN 2025
Aktualizace AISLP říjen 2025 je připravena ke stažení.
Vážení uživatelé programu AISLP, přinášíme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální říjnové verzi AISLP.
Zároveň bychom Vás chtěli upozornit, že pro bezproblémové a automatické stahování měsíčních aktualizací je potřeba program nejen pravidelně používat, ale také musí mít stanice, kde je program nainstalován, přístup k internetu.
Adcetris 50 mg prášek pro koncentrát pro infuzní roztok (brentuximab vedotin) – konjugát monoklonální protilátky a cytostatika – rozšíření indikací, nově: léčba dospělých pacientů s CD30+ HL stadia IIB s rizikovými faktory, stadia III nebo stadia IV v kombinaci s etoposidem, cyklofosfamidem, doxorubicinem, dakarbazinem, dexamethasonem (BrECADD).
Baqsimi 3 mg nosní zásyp v jednodávkovém obalu (glukagon) – hormon – rozšíření indikací, nově u dětí od 1 roku, dříve od 4 let: léčba závažné hypoglykemie u dospělých, dospívajících a dětí od 1 roku s onemocněním diabetes mellitus.
Doritri Máta 0,5 mg/1 mg/1,5 mg pastilky
Doritri Lesní ovoce 0,5 mg/1 mg/1,5 mg pastilky (tyrothricin, benzalkonium-chlorid, benzokain) – podpůrná léčba mírných až středně závažných zánětů faryngu a ústní dutiny u dospělých a dospívajících od 12 let.
mResvia injekční disperze v předplněné injekční stříkačce, mRNA vakcína proti respiračnímu syncytiálnímu viru – rozšíření indikací: k aktivní imunizaci k prevenci onemocnění dolních cest dýchacích (lower respiratory tract disease, LRTD) způsobeného respiračním syncytiálním virem (RSV): u dospělých ve věku 60 let a starších; u dospělých ve věku 18 až 59 let, u nichž je zvýšené riziko onemocnění LRTD způsobeného RSV. Vakcína se má používat v souladu s oficiálními doporučeními.
Pemiros 10 mg/4 mg/1,25 mg, 20 mg/4 mg/1,25 mg, 10 mg/8 mg/2,5 mg a 20 mg/8 mg/2,5 mg potahované tablety (rosuvastatin, perindopril-erbumin, indapamid) – nová fixní kombinace: k prevenci závažných kardiovaskulárních příhod jako náhrada terapie u dospělých pacientů s hypercholesterolemií a esenciální hypertenzí, u kterých jsou tyto rizikové faktory dostatečně kontrolovány perindoprilem/indapamidem užívaným jako dvousložková léková forma a rosuvastatinem jako jednosložkovou lékovou formou, podávanými současně ve stejné dávce jako v kombinaci, ale jako samostatné tablety.
Rezdiffra 60 mg, 80 mg, 100 mg potahované tablety (resmetirom) – hepatoprotektivum – léčba dospělých s necirhotickou steatohepatitidou asociovanou s metabolickou dysfunkcí (MASH) se středně pokročilou až pokročilou jaterní fibrózou (stadium fibrózy F2 až F3).
Riulvy 174 mg a 348 mg enterosolventní tvrdé tobolky (tegomil-fumarát) – léčivo nervového systému – léčba dospělých a pediatrických pacientů ve věku 13 let a starších s relabující-remitující roztroušenou sklerózou.
Tevimbra 100 mg koncentrát pro infuzní roztok (tislelizumab) – cytostatikum, monoklonální protilátka – rozšíření indikací, nově: v kombinaci s chemoterapií obsahující platinu v neoadjuvantní léčbě a následně v monoterapii v adjuvantní léčbě k léčbě resekovatelného nemalobuněčného karcinomu plic (NSCLC) s vysokým rizikem recidivy u dospělých; v kombinaci s gemcitabinem a cisplatinou jako léčba první linie u dospělých pacientů s recidivujícím nazofaryngeálním karcinomem (NPC), u kterého nelze provést kurativní chirurgický zákrok ani radioterapii, nebo metastazujícím NPC.
Tryngolza 80 mg injekční roztok v předplněném peru (olezarsen) – hypolipidemikum, antisense oligonukleotid – doplněk diety u dospělých pacientů k léčbě geneticky potvrzeného syndromu familiární chylomikronemie (familial chylomicronemia syndrome, FCS).
Verdye 5 mg/ml prášek pro injekční roztok (indokyaninová zeleň) – diagnostikum – rozšíření indikací, nově: peroperační identifikace sentinelových lymfatických uzlin a zobrazení lymfatických cév u karcinomu prsu.
Voranigo 10 mg, 40 mg potahované tablety (vorasidenib) – cytostatikum – monoterapie převážně postkontrastně se nesytícího (non-enhancing) astrocytomu nebo oligodendrogliomu 2. stupně s mutací IDH1 R132 nebo IDH2 R172 u dospělých a dospívajících pacientů od 12 let s tělesnou hmotností minimálně 40 kg, kteří podstoupili pouze chirurgický zákrok a bezprostředně nepotřebují radioterapii nebo chemoterapii.
Yeytuo 464 mg injekční roztok (lenakapavir) – antivirotikum – injekce přípravku jsou indikovány v kombinaci s bezpečnějšími sexuálními praktikami k preexpoziční profylaxi (PrEP) ke snížení rizika sexuálním přenosem získané infekce HIV-1 u dospělých a dospívajících s t.hm. nejméně 35 kg, u kterých je zvýšené riziko infekce virem HIV-1.
Yeytuo 300 mg potahované tablety (lenakapavir) – antivirotikum – tablety přípravku jsou indikovány v kombinaci s bezpečnějšími sexuálními praktikami k preexpoziční profylaxi (PrEP) ke snížení rizika sexuálním přenosem získané infekce HIV-1 u dospělých a dospívajících s t.hm. nejméně 35 kg, u kterých je zvýšené riziko infekce virem HIV-1, pro: perorální zahajovací dávku; perorální překlenovací dávku.
Zemcelpro >=0.23 milionů životaschopných buněk CD34+/ml / >=0.53 milionů životaschopných buněk CD3+/ml infuzní disperze – léčba dospělých pacientů s hematologickými malignitami vyžadujícími alogenní transplantaci hematopoetických kmenových buněk po myeloablativním přípravném režimu, pro které není k dispozici žádný jiný typ vhodných dárcovských buněk.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
8.10.2025
Kontrola dávkování léků
O našem projektu si můžete přečíst v článku na Medical Tribune.
Kontrola dávkování léků nyní v článku na Medical Tribune
Kontrola dávkování léků je společný projekt, na kterém se podílí firma INPHARMEX, Farmaceutická fakulta UK Hradec Králové a společnost STAPRO. Funkcionalita "Kontrola dávkování léků" je již nyní součástí nemocničního informačního systému FONS Enterprise společnosti STAPRO.
Nyní si o tomto projektu můžete přečíst v následujícím článku:
Kvůli špatné medikaci ročně umírají statisíce pacientů. Jak tomu zabránit?
Více informací o Kontrole dávkování léků naleznete i v naší Studovně.
Vás tým AISLP
www.aislp.cz
15.9.2025
Moduly AISLP Duplicity a KiK nově v lékárenských systémech (LEKIS a MEDIOX)
Moduly AISLP Duplicity a KiK jsou nyní k dispozici pro uživatele systémů lékárenských softwarů jako je LEKIS a MEDIOX.
Vyzkoušejte si nové moduly Duplicity a KiK.
Duplicity
Modul AISLP Duplicity hledá možné duplicity (i multiplicity) při výdeji léčivých přípravků, tj. zda pacient neužívá dva či více léků se stejnou nebo příbuznou léčivou látkou. Tyto duplicity nehledá jen podle ATC skupiny, ale i podle složení přípravku. Bere tak v úvahu i možnost existence více variant léčivé látky. Modul AISLP Duplicity spolehlivě najde léčivou látku ve všech přípravcích, ve kterých se vyskytuje, tedy i v kombinovaných přípravcích a různých lékových formách.
KiK
KiK slouží jako rychlé upozornění uživatele na možné kontraindikované kombinace léčivých přípravků, které jsou mu předány Vaším lékárenským/ambulantním softwarem formou SÚKL kódů. Modul vychází z informací uvedených v SmPC léčivých přípravků a rychle prověří možné kontraindikace zadaných léčivých přípravků, ať už přímo při výdeji v lékárně, předepisování v ordinaci lékaře nebo při výběru léčivých přípravků z historie pacienta. KiK doplňuje AISLP a umožňuje zobrazit grafický výstup přímo z Vašeho softwaru.
Pro využití těchto funkcí je potřeba mít parametrické volání AISLP předem zapracováno do Vašeho lékárenského softwaru - ověřte si prosím nastavení u Vašeho poskytovatele lékárenského softwaru.
Více o možnostech modulů Duplicity a KiK včetně příkladů naleznete v naší Studovně.
A nyní nabídka pro nové uživatele programu AISLP - vyzkoušejte si KIK a duplicity za výhodných podmínek.
Pokud si objednáte licenci AISLP do konce roku 2025, zaplatíte za každý měsíc do konce roku 2025 jen 1 Kč. Objednejte co nejdříve, ať plně využijete tuto speciální akci.
Tato akce se týká nových uživatelů programu AISLP. Akceptací služby vzniká závazek na 24 měsíců. Akce trvá od 15.9.2025 do 31.12.2025 včetně.
Pro účast na této akci je nutno se registrovat.
V případě zájmu nás neváhejte kontaktovat na tel. 777 746 490 nebo na emailu: info@aislp.cz.
Váš tým AISLP
www.aislp.cz
12.9.2025
Aktualizace ZÁŘÍ 2025
Aktualizace AISLP září 2025 je připravena ke stažení.
Vážení uživatelé programu AISLP,
přinášíme Vám první podzimní výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální zářijové verzi AISLP.
Aucatzyl 10 x 10 na 6 buněk infuzní disperze (obekabtagen autoleucel) – cytostatikum – léčba dospělých pacientů od 26 let s relabující nebo refrakterní akutní lymfoblastickou leukemií z prekurzorů B-lymfocytů (B ALL).
Comirnaty LP.8.1 30 mikrogramů/dávku injekční disperze/injekční disperze v předplněné injekční stříkačce, mRNA vakcína proti onemocnění covid-19 (mRNA kódující kódující spike protein viru SARS-CoV-2 - LP.8.1) – k aktivní imunizaci k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2 u osob ve věku 12 let a starších.
Comirnaty LP.8.1 10 mikrogramů/dávku injekční disperze, mRNA vakcína proti onemocnění covid-19 (mRNA kódující kódující spike protein viru SARS-CoV-2 - LP.8.1) – k aktivní imunizaci k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2 u dětí ve věku 5 až 11 let.
Comirnaty LP.8.1 3 mikrogramy/dávku koncentrát pro injekční disperzi, mRNA vakcína proti onemocnění covid-19 (mRNA kódující kódující spike protein viru SARS-CoV-2 - LP.8.1) – k aktivní imunizaci k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2 u kojenců a dětí ve věku od 6 měsíců do 4 let.
Vakcínu je třeba používat v souladu s oficiálními doporučeními.
Cyclopentanolate Olikla 10 mg/ml oční kapky, roztok (cyklopentolát-hydrochlorid) – oftalmologikum –dilatace zornice při oftalmoskopii a/nebo cykloplegie při refrakčním vyšetření oka; k prevenci a/nebo léčbě synechií při iritidě, iridocyklitidě, keratitidě a choroiditidě.
Maapliv infuzní roztok – roztok aminokyselin bez aminokyselin s rozvětveným řetězcem k léčbě nemoci javorového sirupu (MSUD), která se projevuje akutní dekompenzační epizodou u pacientů, kteří nejsou způsobilí k podávání perorálního a enterálního přípravku bez aminokyselin s rozvětveným řetězcem. Pro všechny věkové skupiny.
Ogsiveo 50 mg, 100 mg, 150 mg potahované tablety (nirogacestat-dihydrobromid) – cytostatikum – monoterapie dospělých pacientů s progredujícími desmoidními nádory, kteří vyžadují systémovou léčbu.
Rezolsta 800 mg/150 mg a 75 mg/150 mg potahované tablety (darunavir, kobicistat) – nová síla přípravku a rozšíření indikace: v kombinaci s dalšími antiretrovirovými léčivými přípravky k léčbě dospělých a pediatrických pacientů (ve věku 6 let a starších s t.hm. nejméně 25 kg) s infekcí způsobenou virem lidské imunodeficience 1 (HIV-1). K nasazení přípravku mají vést genotypové testy.
Sarclisa 20 mg/ml koncentrát pro infuzní roztok (izatuximab) – cytostatikum – rozšíření indikací, nově: v kombinaci s bortezomibem, lenalidomidem a dexamethasonem k indukční léčbě dospělých pacientů s nově diagnostikovaným mnohočetným myelomem, kteří jsou vhodní k autologní transplantaci kmenových buněk.
Saxenda 6 mg/ml injekční roztok v předplněném peru (liraglutid) – GLP-1 agonista – rozšíření indikací, nově: u dětí (6 až < 12 let) je přípravek indikován jako doplňková léčba ke zdravé výživě a zvýšené fyzické aktivitě k úpravě tělesné hmotnosti u dětí ve věku od 6 do < 12 let s:
- obezitou (BMI >= 95. percentil) a
- tělesnou hmotností >= 45 kg.
Pokud u pacientů při dávce 3,0 mg/den nebo při maximální tolerované dávce nedojde po 12 týdnech k poklesu jejich BMI nebo BMI z-skóre alespoň o 4%, léčba přípravkem má být přerušena a přehodnocena.
Skyrizi 180 mg injekční roztok v zásobní vložce (risankizumab) – nová síla přípravku.
Spevigo 300 mg injekční roztok v předplněné injekční stříkačce (spesolimab) – nová síla přípravku.
Spikevax LP.8.1 0,1 mg/ml injekční disperze
Spikevax LP.8.1 50 mikrogramů injekční disperze v předplněné injekční stříkačce
mRNA vakcína proti onemocnění covid-19 (mRNA kódující kódující spike protein viru SARS-CoV-2 - LP.8.1) – k aktivní imunizaci jedinců od 6 měsíců věku k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2. Vakcínu je třeba používat v souladu s oficiálními doporučeními.
Xofluza 10 mg, 30 mg a 40 mg granule v sáčku baloxavir marboxil – antivirotikum – nová léková forma přípravku: k léčbě nekomplikované chřipky u pacientů ve věku od 3 týdnů; k postexpoziční profylaxi chřipky u jedinců ve věku od 3 týdnů. Přípravek je třeba používat v souladu s oficiálními doporučeními.
Veklury 100 mg prášek pro koncentrát pro infuzní roztok (remdesivir) – antivirotikum – rozšíření indikací: k léčbě onemocnění covid-19 u dospělých a pediatrických pacientů (ve věku nejméně 4 týdny s t.hm. nejméně 3 kg): s pneumonií vyžadující doplňkovou oxygenoterapii (kyslík o nízkém nebo vysokém průtoku nebo jinou neinvazivní ventilaci na začátku léčby); u pacientů, kteří nevyžadují doplňkovou oxygenoterapii a u nichž je zvýšené riziko progrese do závažného onemocnění covid-19.
Wezenla 90 mg a 45 mg injekční roztok v předplněné injekční stříkačce a 45 mg injekční roztok (ustekinumab) – rozšíření indikací pro pediatrické pacienty s t.hm. od 40 kg: léčba pediatrických pacientů se středně těžkou až těžkou aktivní Crohnovou chorobou s t.hm. nejméně 40 kg, u kterých buď odpověď na konvenční terapii nebo na biologickou terapii nebyla dostatečná nebo tito pacienti konvenční nebo biologickou léčbu netolerovali.
Wezenla 130 mg koncentrát pro infuzní roztok (ustekinumab) – rozšíření indikací na pediatrické pacienty s t.hm. od 40 kg: léčba pediatrických pacientů se středně těžkou až těžkou aktivní Crohnovou chorobou s t.hm. nejméně 40 kg, u kterých buď odpověď na konvenční terapii nebo na biologickou terapii nebyla dostatečná nebo tito pacienti konvenční nebo biologickou léčbu netolerovali.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
1.9.2025
Aktualizované obchodní podmínky
Aktualizovali jsme obchodní podmínky.
Od 1.9.2025 došlo k aktualizaci a rozšíření o ustanovení týkající se podmínek marketinkových akcí a z nich plynoucích závazků.
Aktuální platné znění naleznete pod odkazem: Aktuální obchodní podmínky.
Váš tým AISLP
www.aislp.cz
18.8.2025
Aktualizace SRPEN 2025
Aktualizace AISLP srpen 2025 je připravena ke stažení.
Vážení uživatelé programu AISLP,
přinášíme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální srpnové verzi AISLP.
Adempas 0,5 mg, 1 mg, 1,5 mg, 2 mg a 2,5 mg potahované tablety
Adempas 0,15 mg/ml granule pro perorální suspenzi (riociguát) – rozšíření indikací a nová léková forma pro pediatrické pacienty: v kombinaci s antagonisty receptoru pro endotelin k léčbě plicní arteriální hypertenze (PAH) u pediatrických pacientů ve věku od 6 do 18 let s funkční třídou II až III dle WHO.
Alyftrek 50 mg/20 mg/4 mg potahované tablety
Alyftrek 125 mg/50 mg/10 mg potahované tablety (deutivakaftor, tezakaftor, dihydrát vápenaté soli vanzakaftoru) – léčba cystické fibrózy u pacientů ve věku od 6 let, kteří mají alespoň jednu mutaci v genu CFTR jiné třídy než třídy I.
Attrogy 250 mg potahované tablety (diflunisal) – léčba hereditární transthyretinové amyloidózy (hATTR) u dospělých pacientů s polyneuropatií v 1. či 2. stádiu (hATTR-PN).
Ezmekly 1 mg a 2 mg tvrdé tobolky, 1 mg dispergovatelné tablety (mirdametinib) – cytostatikum – monoterapie symptomatických, neoperovatelných plexiformních neurofibromů u dospělých a pediatrických pacientů od 2 let s neurofibromatózou typu 1 (NF1).
Efluelda injekční suspenze v předplněné injekční stříkačce, trivalentní vakcína proti chřipce
Efluelda Tetra injekční suspenze v předplněné injekční stříkačce, tetravalentní vakcína proti chřipce (štěpený virion, inaktivovaný), 60 mikrogramů HA/kmen
Fluad injekční suspenze v předplněné injekční stříkačce, vakcína proti chřipce (povrchový antigen, inaktivovaná, adjuvovaná)
Fluarix Tetra injekční suspenze v předplněné injekční stříkačce, vakcína proti chřipce (štěpený virion, inaktivovaný)
Flucelvax injekční suspenze v předplněné injekční stříkačce, vakcína proti chřipce (povrchový antigen, inaktivovaná, připravená na buněčných kulturách)
Influvac injekční suspenze v předplněné injekční stříkačce
Influvac Tetra injekční suspenze v předplněné injekční stříkačce, vakcína proti chřipce (povrchový antigen, inaktivovaná) aktualizace složení vakcín proti sezónní chřipce podle doporučení Světové zdravotnické organizace (WHO) pro severní polokouli a doporučení Evropské unie pro sezónu 2025/2026.
Fluenz nosní sprej, suspenze, vakcína proti chřipce (živá, nosní), aktualizace složení pro sezónu 2025/2026, aktualizace SmPC a PIL.
Itovebi 3 mg a 9 mg potahované tablety (inavolisib) – cytostatikum – v kombinaci s palbociklibem a fulvestrantem k léčbě dospělých pacientů s estrogen-receptor (ER) pozitivním, HER2 negativním, lokálně pokročilým nebo metastazujícím karcinomem prsu s mutací genu PIK3CA, po relapsu během adjuvantní endokrinní léčby nebo do 12 měsíců od jejího dokončení.
Jivi 4000 IU prášek a rozpouštědlo pro injekční roztok (damoctokog alfa pegol) – nová síla k léčbě a profylaxi krvácení u pacientů s hemofilií A.
Labrycor 0,2 mg/ml koncentrát pro infuzní roztok (isoprenalin-hydrochlorid) – sympatomimetikum – změna lékové formy a aktualizace SmPC: krátkodobá léčba trvalé bradykardie způsobené atrio-ventrikulární blokádou v době, kdy se čeká na kardiostimulátor nebo kdy je kardiostimulátor kontraindikován. Krátkodobá léčba Adamsova-Stokesova syndromu. Je třeba dodržovat národní a mezinárodní doporučení a pokyny týkající se vhodného použití isoprenalinu.
Nurofen prolong 300 mg tablety s prodlouženým uvolňováním (ibuprofen) – nová léková forma přípravku – nesteroidní antiflogistikum – krátkodobá léčba mírné až středně silné bolesti, u které se očekává, že bude trvat déle než 6-8 hodin, jako je bolest zad, bolest svalů, bolest kloubů, menstruační bolest a bolest zubů. Pouze pro dospělé.
Omlyclo 75 mg injekční roztok v předplněném peru
Omlyclo 150 mg injekční roztok v předplněném peru (omalizumab) – nová léková forma pro pacienty od 12 let.
Osirid 5 mg/ml injekční roztok (sodná sůl torasemidu) – diuretikum – léčba kardiálních edémů a/nebo výpotků v důsledku srdečního selhání, pokud je nutná intravenózní terapie (např. plicní edém v důsledku akutního srdečního selhání). Přípravek je indikován k léčbě dospělých.
Ziihera 300 mg prášek pro koncentrát pro infuzní roztok (zanidatamab) – cytostatikum – monoterapie dospělých pacientů s neresekovatelným lokálně pokročilým nebo metastazujícím HER2-pozitivním (IHC 3+) karcinomem žlučových cest (BTC), kteří byli dříve léčeni alespoň jednou linií systémové terapie.
Zoonotic Influenza Vaccine Seqirus injekční suspenze v předplněné injekční stříkačce, vakcína proti zoonotické chřipce (H5N8) (povrchový antigen, inaktivovaná, s adjuvans) – rozšíření indikací: k aktivní imunizaci proti subtypu H5 viru chřipky typu A u jedinců ve věku 6 měsíců a starších. Použití vakcíny má být v souladu s oficiálními doporučeními.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
8.8.2025
Otestujte si kontrolu dávkování léků
Společný projekt AISLP a STAPRO nyní zdarma k vyzkoušení.
Kontrola dávkování léků je společný projekt, na kterém se podílí firma INPHARMEX, Farmaceutická fakulta UK Hradec Králové a společnost STAPRO. Funkcionalita "Kontrola dávkování léků" je již nyní součástí nemocničního informačního systému FONS Enterprise společnosti STAPRO.
Stále běží akce pro nemocnice
Až do poloviny září si můžete zdarma vyzkoušet a otestovat funkcionalitu Kontrola dávkování léků.
V případě, že již testujete, nebo uvažujete o zahájení testování, nabízíme Vám k tomu nyní možnost pořídit si plnou licenci AISLP se slevou 50 % na prvních 12 měsíců.
Tato nabídka se týká pouze zakoupení nové plné licence AISLP při souběžném nasazení modulu Kontrola dávkování léků.
Optimální fungování Kontroly dávkování léků
Pořízení licence AISLP vám umožní využívat další detailní textové informace přímo ve FONS ENTERPRISE, například kliknutím na ikonu SmPC otevřít informace o dávkování v systému AISLP.
Jak funguje Kontrola dávkování léků
Tato funkcionalita automaticky kontroluje dávky léků ordinovaných ve strukturované medikaci a receptu dle hodnot maximální, doporučené nebo obvyklé dávky v SmPC daného léku s ohledem na základní kritéria pacienta.
FONS Enterprise provádí vyhodnocení předaných informací o maximální nebo doporučené dávce léku s dávkováním v ordinaci léku a zobrazuje upozornění v případech, kdy ordinované dávky nejsou v souladu s doporučením nebo nemohou být vyhodnoceny.
Ordinující lékař je při překročení dávky, nevhodném či nedoporučeném dávkování v ordinaci, informován formou různého barevného zabarvení rohu položky Rozpis/Dávka a doplňkovou zkrácenou textovou informací o dávkování z SmPC.
Obdobně zde funguje možnost přímého prokliku do systému AISLP do části SmPC léku „4.2 Dávkování“
Více informací o Kontrole dávkování léků naleznete v naší Studovně.
V případě zájmu o tento produkt nás prosím kontaktujte telefonicky: 777 746 490, nebo emailem: info@aislp.cz.
Váš tým AISLP
www.aislp.cz
22.7.2025
Novinky v AISLP
Moduly Duplicity a KiK v databázi AISLP.
Vážení předplatitelé databáze léků AISLP,
jistě jste již zaznamenali některé novinky v databázi AISLP. Spolu s červencovou aktualizací máte možnost začít využívat moduly Duplicity a KiK, které jsou rozšířením základní verze databáze AISLP.
Modul AISLP Duplicity hledá možné duplicity (i multiplicity) při výdeji léčivých přípravků, tj. zda pacient neužívá dva či více léků se stejnou nebo příbuznou léčivou látkou.
Grafické zobrazení umožňuje rychlou a vizuální kontrolu případných duplicit.
Modul AISLP KiK slouží jako rychlé signální upozornění při výdeji léčivých přípravků na možné závažné kontraindikace. Výsledky jsou zobrazeny graficky a ukazují počet nalezených kontraindikovaných kombinací včetně jejich krátkého popisu.
Funkčnost uvedených modulů závisí na verzi používaného lékárenského softwaru. Ověřte si u svého dodavatele lékárenského systému, zda je možné tyto moduly u Vás použít.
Věříme, že se oba tyto moduly brzy stanou Vaším nepostradatelným pomocníkem při práci s databází AISLP.
Více o možnostech duplicit a KiK naleznete na www.aislp.cz.
Váš tým AISLP
www.aislp.cz
16.7.2025
Aktualizace ČERVENEC 2025
Aktualizace AISLP červenec 2025 je připravena ke stažení.
Vážení uživatelé programu AISLP,
přinášíme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální červencové verzi AISLP.
Spolu s červencovou aktualizací máte možnost začít využívat moduly Duplicity a KiK (kontraindikované kombinace), které jsou rozšířením základní verze databáze AISLP.
Více o těchto modulech a možnostech používání v závislosti na vašem lékárenském systému naleznete na www.aislp.cz.
Amvuttra 25 mg injekční roztok v předplněné injekční stříkačce (vutrisiran) – rozšíření indikací, nově: léčba získané (wild-type, wtATTR) nebo hereditární transthyretinové amyloidózy u dospělých pacientů s kardiomyopatií (ATTR-CM).
Breyanzi 1,1-70 x 10 na 6 buněk/ml infuzní disperze (lisokabtagen maraleucel) – cytostatikum – rozšíření indikací, nově: léčba dospělých pacientů s relabujícím nebo refrakterním folikulárním lymfomem (FL) po dvou nebo více liniích systémové terapie.
Bronchipret tymián a břečťan perorální roztok (tekutý extrakt z tymiánu a břečťanového listu) – nová léková forma k použití jako expektorans při produktivním kašli u dospělých a dospívajících od 12 let.
Duvyzat 8,86 mg/ml perorální suspenze (givinostat) – léčivo pro poruchy muskuloskeletálního systému – léčba Duchennovy svalové dystrofie (DMD) u ambulantních (chodících) pacientů ve věku 6 let a starších a při současné léčbě kortikosteroidy.
Evrysdi 5 mg potahované tablety (risdiplam) – nová léková forma přípravku – léčba spinální svalové atrofie (SMA) vázané na dlouhé raménko 5. chromozomu u pacientů s klinickou diagnózou SMA 1. typu, 2. typu nebo 3. typu nebo s jednou až čtyřmi kopiemi genu SMN2.
Framykoin 3300 IU/g + 250 IU/g mast (neomycin-sulfát, zinečnatý komplex bacitracinu) – změna vyjádření síly (bez změny obsahu léčivých látek v přípravku, dříve Framykoin 250 IU/g + 5,2 mg mast).
Itulazax 12 SQ-Bet sublingvální lyofilizát (standardizovaný alergenový extrakt z pylu břízy bradavičnaté, Betula verrucosa) – přípravek pro alergenovou imunoterapii – rozšíření indikace pro děti od 5 let: k léčbě dospělých a dětí (od 5 let) se středně závažnou až závažnou alergickou rinitidou a/nebo konjuktivitidou vyvolanou pylem ze skupiny alergenů homologních s břízou (pyly břízy, olše, lísky, habru, dubu a buku).
Jivi 250 IU, 500 IU, 1000 IU 2000 IU a 3000 IU prášek a rozpouštědlo pro injekční roztok (damoctokog alfa pegol) – rozšíření indikace pro děti od 7 let: léčba a profylaxe krvácení u dříve léčených pacientů ve věku >=7 let s hemofilií A (vrozený nedostatek faktoru VIII).
Ophthalmo-Framykoin 3300 IU/g + 250 IU/g oční mast (neomycin-sulfát, zinečnatý komplex bacitracinu) – změna vyjádření síly (bez změny obsahu léčivých látek v přípravku, dříve Ophthalmo-Framykoin 250 IU/g + 5,2 mg/g oční mast).
Ophthalmo-Framykoin comp. 3300 IU/g + 250 IU/g + 5 mg/g oční mast (neomycin-sulfát, zinečnatý komplex bacitracinu, hydrokortison-acetát) – změna vyjádření síly (bez změny obsahu léčivých látek v přípravku, dříve Ophthalmo-Framykoin comp. 250 IU/g + 5,2 mg/g + 5 mg/g oční mast).
Ronapreve 300 mg + 300 mg injekční/infuzní roztok
Ronapreve 1332 mg + 1332 mg injekční/infuzní roztok (kasirivimab, imdevimab) – antivirové monoklonální protilátky – rozšíření indikace pro děti od 2 let: k léčbě onemocnění covid-19 u dospělých, dospívajících a dětí ve věku 2 let a starších s tělesnou hmotností nejméně 10 kg, kteří nevyžadují oxygenoterapii a kteří mají zvýšené riziko progrese do těžké formy onemocnění covid-19.
Ryjunea 0,1 mg/ml oční kapky, roztok (atropin-sulfát) – oftalmologikum, mydriatikum – zpomalení progrese myopie u dětí a dospívajících ve věku 3-14 let s roční mírou progrese 0,50 dioptrie (D) a více a se závažností -0,5 D až -6,0 D.
Sarclisa 20 mg/ml koncentrát pro infuzní roztok (izatuximab) – cytostatikum, monoklonální protilátka – rozšíření indikací, nově: v kombinaci s bortezomibem, lenalidomidem a dexamethasonem k léčbě dospělých pacientů s nově diagnostikovaným mnohočetným myelomem, kteří nejsou vhodní k autologní transplantaci kmenových buněk.
Sephience 250 mg, 1000 mg perorální prášek v sáčku (sepiapterin) – trávicí trakt a metabolismus - léčba hyperfenylalaninemie (HPA) u dospělých a pediatrických pacientů s fenylketonurií (PKU).
Sivextro 200 mg prášek pro koncentrát pro infuzní roztok (tedizolid-fosfát) – antibiotikum ze skupiny oxazolidinonů – rozšíření indikace: léčba akutních bakteriálních infekcí kůže a kožních struktur (ABSSSI) u dospělých, dospívajících a dětí od narození.
Sivextro 200 mg potahované tablety (tedizolid-fosfát) – antibiotikum ze skupiny oxazolidinonů – rozšíření indikace: léčba akutních bakteriálních infekcí kůže a kožních struktur (ABSSSI) u dospělých, dospívajících a dětí s t.hm. alespoň 35 kg.
Tepezza 500 mg prášek pro koncentrát pro infuzní roztok (teprotumumab) – imunosupresivum, monoklonální protilátka – léčba dospělých se středně závažnou až závažnou endokrinní orbitopatií (EO).
Tevimbra 100 mg koncentrát pro infuzní roztok (tislelizumab) – cytostatikum, monoklonální protilátka – rozšíření indikací, nově: v kombinaci s etoposidem a chemoterapií obsahující platinu jako první linie léčby dospělých pacientů v extenzivním stádiu malobuněčného karcinomu plic (SCLC).
Vaxigrip injekční suspenze v předplněné injekční stříkačce, trivaletntní vakcína proti chřipce
Vaxigrip Tetra injekční suspenze v předplněné injekční stříkačce, tetravalentní vakcína proti chřipce (štěpený virion, inaktivovaný) – aktualizace složení vakcín proti sezónní chřipce dle doporučení Světové zdravotnické organizace (WHO) pro severní polokouli a doporučení Evropské unie pro sezónu 2025/2026.
Vyvgart 1000 mg injekční roztok v předplněné injekční stříkačce (efgartigimod alfa) – selektivní imunosupresivum, nová léková forma přípravku – přídatná terapie ke standardní léčbě dospělých pacientů s generalizovanou myasthenia gravis (gMG), kteří mají pozitivní nález protilátek proti acetylcholinovým receptorům (AChR).
Xados 2,5 mg/ml perorální roztok
Xados 10 mg tablety dispergovatelné v ústech (bilastin) – rozšíření indikace pro děti od 2 let: symptomatická léčba alergické rinokonjunktivitidy (sezónní i celoroční) a kopřivky u dětí ve věku 2 až 11 let s t.hm. alespoň 15 kg.
Xoanacyl 1 g potahované tablety (koordinační komplex ferrum(III)-citrátu) – přípravek k léčbě hyperfosfatemie a nedostatku železa – léčba zvýšených hladin fosforu v séru a souběžného nedostatku železa u dospělých pacientů s chronickým onemocněním ledvin.
Xofluza 20 mg, 40 mg, 80 mg potahované tablety
Xofluza 2 mg/ml granule pro perorální suspenzi (baloxavir marboxil) – antivirotikum – rozšíření indikace pro děti od 3 týdnů věku: léčba nekomplikované chřipky u pacientů ve věku od 3 týdnů; postexpoziční profylaxe chřipky u jedinců ve věku od 3 týdnů.
Xydalba 500 mg prášek pro koncentrát pro infuzní roztok (dalbavancin) – antibiotikum ze skupiny glykopeptidů – rozšíření indikace: léčba akutních bakteriálních infekcí kůže a kožních struktur (ABSSSI) u dospělých a pediatrických pacientů od narození.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
30.6.2025
Letní akce 2025
Vyražte na prázdniny s AISLP!
Využijte letní akci a pořiďte si AISLP v termínu od 1.7. do 31.8.2025.
Stačí jen kliknout na banner a vyplnit poptávkový formulář.

Přejeme Vám krásné léto!
Váš tým AISLP
www.aislp.cz
13.6.2025
Aktualizace ČERVEN 2025
Aktualizace AISLP červen 2025 je připravena ke stažení.
Vážení uživatelé programu AISLP,
dříve než začne léto a čas dovolených, přinášíme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální červnové verzi AISLP.
Aqumeldi 1 mg tablety dispergovatelné v ústech (enalapril-maleinát) – nová síla přípravku: k léčbě srdečního selhání u dětí od narození do méně než 18 let.
Blincyto 38,5 mikrogramu prášek pro koncentrát pro infuzní roztok (blinatumomab) – cytostatikum – rozšíření indikací, nově: monoterapie v rámci konsolidační léčby dospělých pacientů s nově diagnostikovanou Philadelphia chromozom negativní CD19 pozitivní ALL z prekurzorů B buněk; použití u dětí od 1 měsíce (dříve od 1 roku) v léčbě Philadelphia chromozom negativní CD19 pozitivní ALL z prekurzorů B buněk.
Cedepos 125 mg tvrdé tobolky (vankomycin-hydrochlorid) – přípravek je indikován u pacientů od 12 let k léčbě infekce způsobené Clostridioides difficile (CDI). Je nutno vzít v úvahu oficiální doporučení ke správnému používání antibakteriálních léčiv.
Dagrafors Duo 100 mg/10 mg potahované tablety (sitagliptin a dapagliflozin, ve formě monohydrátu sitagliptin-fosfátu a monohydrátu dapagliflozin-propandiolu) – perorální antidiabetikum, kombinace – přípravek je indikován u dospělých ve věku 18 let a starších k léčbě diabetes mellitus 2. typu jako doplněk k dietě a cvičení jako náhrada terapie u pacientů adekvátně kontrolovaných dapagliflozinem a sitagliptinem podávanými současně ve stejné dávce jako v kombinaci, ale jako samostatné tablety.
Lynozyfic 5 mg a 200 mg koncentrát pro infuzní roztok (linvoseltamab) – cytostatikum, monoklonální protilátka – monoterapie dospělých pacientů s relabujícím a refrakterním mnohočetným myelomem, kteří dostali nejméně 3 předchozí terapie zahrnující inhibitor proteazomu, imunomodulační přípravek a monoklonální protilátku proti CD38, a při poslední terapii vykázali progresi onemocnění.
Lyrica 25 mg, 75 mg a 150 mg tablety dispergovatelné v ústech (pregabalin) – antiepileptikum, analgetikum – nová léková forma přípravku: léčba periferní a centrální neuropatické bolesti u dospělých. Přídatná léčba epilepsie u dospělých s parciálními záchvaty se sekundární generalizací nebo bez ní. Léčba generalizované úzkostné poruchy u dospělých.
Spikevax XBB.1.5 25 mikrogramů injekční disperze v předplněné injekční stříkačce, mRNA vakcína proti onemocnění covid-19 (andusomeran) – k aktivní imunizaci jedinců od 6 měsíců věku k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2. Vakcínu je třeba používat v souladu s oficiálními doporučeními.
Torasemid Stada 2,5 mg, 5 mg, 10 mg a 20 mg tablety (torasemid) – diuretikum – 2,5 mg: esenciální hypertenze u dospělých pacientů; 5 mg, 10 mg a 20 mg: léčba a prevence rekurence kardiálního edému a/nebo výpotku u dospělých v důsledku srdečního selhání.
Tremfya 200 mg koncentrát pro infuzní roztok a injekční roztok v předplněné injekční stříkačce/peru (guselkumab) – imunosupresivum, inhibitor interleukinu – nová síla a léková forma přípravku: léčba dospělých pacientů se středně těžkou až těžkou aktivní ulcerózní kolitidou, kteří měli nedostatečnou odpověď, ztratili odpověď nebo netolerovali buď konvenční léčbu, nebo biologickou léčbu. Léčba dospělých pacientů se středně těžkou až těžkou aktivní Crohnovou chorobou, kteří měli nedostatečnou odpověď, ztratili odpověď nebo netolerovali buď konvenční léčbu, nebo biologickou léčbu.
Tremfya 100 mg injekční roztok v předplněné injekční stříkačce/peru (guselkumab) – imunosupresivum, inhibitor interleukinu – rozšíření indikací: léčba středně těžké až těžké plakové psoriázy u dospělých, kteří jsou kandidáty pro systémovou léčbu. V monoterapii nebo v kombinaci s methotrexátem (MTX) k léčbě aktivní psoriatické artritidy u dospělých pacientů, kteří nedostatečně odpovídali na předchozí terapii chorobu modifikujícím antirevmatikem (DMARD) nebo ji netolerovali. Léčba dospělých pacientů se středně těžkou až těžkou aktivní ulcerózní kolitidou, kteří měli nedostatečnou odpověď, ztratili odpověď nebo netolerovali buď konvenční léčbu, nebo biologickou léčbu. Léčba dospělých pacientů se středně těžkou až těžkou aktivní Crohnovou chorobou, kteří měli nedostatečnou odpověď, ztratili odpověď nebo netolerovali buď konvenční léčbu, nebo biologickou léčbu.
Vyjuvek 5 x 109 jednotek tvořících plaky/ml suspenze a gel pro přípravu gelu (beremagen geperpavek) – přípravek pro topickou genovou terapii – k léčbě ran u pacientů s dystrofickou bulózní epidermolýzou s mutací (mutacemi) v genovém řetězci kolagenu typu VII alfa 1 (COL7A1), a to od narození.
Prevymis 240 mg a 480 mg potahované tablety (letermovir) – antivirotikum – rozšíření indikací: k profylaxi reaktivace a rozvoje onemocnění způsobeného cytomegalovirem (CMV) u dospělých a pediatrických pacientů s tělesnou hmotností nejméně 15 kg, kteří jsou CMV séropozitivní příjemci [R+] podstupující alogenní transplantaci hematopoetických kmenových buněk (HSCT). K profylaxi rozvoje onemocnění způsobeného CMV u dospělých a pediatrických pacientů s tělesnou hmotností nejméně 40 kg, kteří jsou CMV séronegativní příjemci transplantované ledviny od CMV séropozitivního dárce [D+/R-]. Je nutno věnovat pozornost oficiálním pokynům ke správnému používání antivirotik.
Prevymis 240 mg a 480 mg koncentrát pro infuzní roztok (letermovir) – antivirotikum – rozšíření indikací: k profylaxi reaktivace a rozvoje onemocnění způsobeného cytomegalovirem (CMV) u dospělých a pediatrických pacientů s tělesnou hmotností nejméně 5 kg, kteří jsou CMV séropozitivní příjemci [R+] podstupující alogenní transplantaci hematopoetických kmenových buněk (HSCT). K profylaxi rozvoje onemocnění způsobeného CMV u dospělých a pediatrických pacientů s tělesnou hmotností nejméně 40 kg, kteří jsou CMV séronegativní příjemci transplantované ledviny od CMV séropozitivního dárce [D+/R-]. Je nutno věnovat pozornost oficiálním pokynům ke správnému používání antivirotik.
Prevymis 20 mg a 120 mg granule v sáčku roztok (letermovir) – antivirotikum – nová síla a léková forma: k profylaxi reaktivace a rozvoje onemocnění způsobeného cytomegalovirem (CMV) u dospělých a pediatrických pacientů s tělesnou hmotností nejméně 5 kg, kteří jsou CMV séropozitivní příjemci [R+] podstupující alogenní transplantaci hematopoetických kmenových buněk (HSCT). K profylaxi rozvoje onemocnění způsobeného CMV u dospělých a pediatrických pacientů s tělesnou hmotností nejméně 40 kg, kteří jsou CMV séronegativní příjemci transplantované ledviny od CMV séropozitivního dárce [D+/R-]. Je nutno věnovat pozornost oficiálním pokynům ke správnému používání antivirotik.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
15.5.2025
Úprava ceny od 1.7.2025
V souladu se stávajícími obchodními podmínkami upravujeme od 1.7.2025 i ceník pro stávající předplatitele.
Vážení předplatitelé databáze léků AISLP,
velice si vážíme Vaší přízně a důvěry, kterou nám projevujete užíváním naší databáze. Bohužel v souvislosti s nárůstem všech nákladů od roku 2022, kdy naposledy došlo ke změně ceny, jsme nuceni také reagovat a přistoupit k její úpravě.
Cenu jsme se rozhodli upravit pouze o minimální výši 5 %. Např. u nejběžnější licence pro 1-2 počítače se jedná o zvýšení o necelých 30 Kč měsíčně a stále je zachováno cenové zvýhodnění oproti novým předplatitelům, kteří se rozhodnou objednat AISLP až během roku 2025.
Abychom Vám změnu ceny částečně kompenzovali, nabízíme Vám možnost rozšíření stávajícího programu AISLP na verzi s pokročilým vyhledáváním v databázi na duplicity a případné „Kontraindikované kombinace“*. Stačí pouze zachovat Vaše předplatné a v závislosti na technických možnostech poskytovatele Vašeho ambulantního a lékárenského softwaru, dojde v červencové aktualizaci k automatickému rozšíření na verzi s těmito možnostmi.
Zároveň i díky zlepšené spolupráci s našimi partnery došlo ke zjednodušení procesu přechodu na rozšířenou verzi s pokročilým vyhledáváním a potvrzení pokračování předplatného.
V případě jakýchkoliv nejasností nás prosím, buď přímo, nebo prostřednictvím našeho partnera, neváhejte kontaktovat.
Věříme, že i přes tuto méně příjemnou zprávu nám zachováte svoji přízeň a těšíme se na další spolupráci.
Váš tým AISLP
*Vice o možnostech duplicit a KiK naleznete na www.aislp.cz
13.5.2025
Aktualizace KVĚTEN 2025
Aktualizace AISLP květen 2025 je připravena ke stažení.
Vážení uživatelé programu AISLP,
v následujícím přehledu Vám přinášíme výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální květnové verzi AISLP.
Abrysvo prášek a rozpouštědlo pro injekční roztok, vakcína proti respiračnímu syncytiálnímu viru, bivalentní, rekombinantní (stabilizovaný prefuzní F antigen RSV podskupiny A a RSV podskupiny B) – rozšíření indikací: k pasivní ochraně před onemocněním dolních cest dýchacích způsobeným respiračním syncytiálním virem (RSV) u kojenců od narození do 6 měsíců věku po imunizaci matky během těhotenství. K aktivní imunizaci osob ve věku 18 let a starších k prevenci onemocnění dolních cest dýchacích způsobeného RSV. Vakcínu je třeba používat v souladu s oficiálními doporučeními.
Capvaxive injekční roztok v předplněné injekční stříkačce, pneumokoková polysacharidová konjugovaná vakcína (21valentní) – k aktivní imunizaci za účelem prevence invazivního onemocnění a pneumonie způsobené Streptococcus pneumoniae u jedinců ve věku 18 let a starších. Použití vakcíny má být v souladu s oficiálními doporučeními.
Datroway 100 mg prášek pro koncentrát pro infuzní roztok (datopotamab deruxtekan) – cytostatikum, konjugát protilátka-léčivo – léčba dospělých pacientů s neresekovatelným nebo metastazujícím HR-pozitivním, HER2-negativním karcinomem prsu, kteří podstoupili endokrinní terapii a alespoň jednu linii chemoterapie v režimu pokročilého karcinomu.
Enhertu 100 mg prášek pro koncentrát pro infuzní roztok (trastuzumab deruxtekan) – cytostatikum, konjugát protilátka-léčivo – rozšíření indikací, nově: v monoterapii k léčbě dospělých pacientů s neresekovatelným nebo metastazujícím karcinomem prsu s pozitivními hormonálními receptory (HR), nízkou hladinou HER2 nebo ultranízkou hladinou HER2, kteří podstoupili alespoň jednu endokrinní léčbu v režimu pro metastazující karcinom a kteří nejsou v další linii léčby považováni za vhodné kandidáty pro endokrinní léčbu
Fabhalta 200 mg tvrdé tobolky (iptakopan) – imunosupresivum, inhibitor komplementu – rozšíření indikací, nově: léčba dospělých pacientů s glomerulopatií C3 složky komplementu (C3G), a to buď v kombinaci s inhibitorem renin-angiotenzinového systému (RAS), nebo samostatně u pacientů s intolerancí inhibitoru RAS nebo u kterých je inhibitor RAS kontraindikován.
Flucelvax injekční suspenze v předplněné injekční stříkačce, vakcína proti chřipce (povrchový antigen, inaktivovaná, připravená na buněčných kulturách) – rozšíření indikace: profylaxe chřipky u dospělých a dětí od 6 měsíců věku. Přípravek má být podáván v souladu s oficiálními doporučeními.
Ixchiq prášek a rozpouštědlo pro injekční roztok, vakcína proti onemocnění Chikungunya (živý oslabený Chikungunya virus (CHIKV), kmen delta5nsP3, produkovaný ve Vero buňkách) – rozšíření indikace: k aktivní imunizaci k prevenci onemocnění způsobeného Chikungunya virem (CHIKV) u osob ve věku 12 let a starších. Vakcínu je třeba používat v souladu s oficiálními doporučeními.
Labrycor 0, 2 mg/ml a 1 mg/5 ml injekční/infuzní roztok (isoprenalin hydrochlorid) – sympatomimetikum – léčivý přípravek pro naléhavé situace: Adamsův-Stokesův syndrom způsobený atrioventrikulární blokádou, zatímco se čeká na zařízení pro dočasné nebo trvalé řešení; extrémní bradykardie se synkopou způsobenou sinoatriální blokádou, zatímco se čeká na zařízení pro dočasné nebo trvalé řešení; srdeční zástava, když se znovu objevuje srdeční aktivita; nízký srdeční výdej po kardiochirurgickém výkonu; torsade de pointes v případě, že není možné při rychlé komorové frekvenci provést stimulaci, spolu s kauzální léčbou torsade de pointes a obnovením normální hladiny draslíku v séru.
Leqembi 100 mg/ml koncentrát pro infuzní roztok (lekanemab) – léčivo proti demenci – léčba dospělých pacientů s klinickou diagnózou mírné kognitivní poruchy a lehké demence způsobené Alzheimerovou chorobou (časná Alzheimerova choroba), kteří nejsou nositeli genu apolipoproteinu E epsilon 4 (ApoE4) nebo jsou heterozygoti s potvrzenou amyloidovou patologií.
Pixoroso 10 mg/4 mg, 10 mg/8 mg, 20 mg/4 mg a 20 mg/8 mg potahované tablety (rosuvastatin, perindopril-erbumin) – nová fixní kombinace: náhrada léčby u dospělých pacientů, kteří jsou adekvátně kontrolováni současným podáváním rosuvastatinu a perindoprilu ve stejné dávce jako v kombinaci k léčbě hypertenze u pacientů, u nichž se odhaduje vysoké riziko první kardiovaskulární příhody (pro prevenci závažných kardiovaskulárních příhod) nebo s jedním z následujících souběžných stavů: primární hypercholesterolemie (typ IIa včetně heterozygotní familiární hypercholesterolemie) nebo smíšená dyslipidemie (typ IIb); homozygotní familiární hypercholesterolemie.
Rxulti 0,25 mg, 0,5 mg, 1 mg, 2 mg, 3 mg a 4 mg potahované tablety (brexpiprazol) – antipsychotikum – rozšíření indikací na pediatrické pacienty: léčba schizofrenie u dospělých a dospívajících ve věku od 13 let.
Slenyto 1 mg a 50 mg tablety s prodlouženým uvolňováním (melatonin) – hypnotikum – nová indikace: léčba insomnie u dětí a dospívajících ve věku 6-17 let s poruchou pozornosti s hyperaktivitou (ADHD), kde nejsou opatření v oblasti hygieny spánku dostatečná.
Supemtek Tetra injekční roztok v předplněné injekční stříkačce – tetravalentní vakcína proti chřipce (rekombinantní, připravovaná na buněčných kulturách) – rozšíření indikace: k aktivní imunizaci k prevenci chřipkového onemocnění u dospělých a dětí od 9 let věku. Vakcína musí být používána v souladu s oficiálními doporučeními.
Taltz 80 mg injekční roztok v předplněném peru, 40 mg a 80 mg injekční roztok v předplněné injekční stříkačce (ixekizumab) – imunosupresivum, inhibitor interleukinu – nová síla přípravku 40 mg vhodná pro dávkování u pediatrických pacientů s t.hm. do 50 kg: léčba středně těžké až těžké ložiskové psoriázy u dospělých, dospívajících a dětí ve věku od 6 let s t.hm. alespoň 25 kg, kteří jsou kandidáty pro systémovou léčbu.
Tevimbra 100 mg koncentrát pro infuzní roztok (tislelizumab) – cytostatikum, monoklonální protilátka – rozšíření indikací, nově: v kombinaci s chemoterapií založenou na platině a fluorpyrimidinu k léčbě první linie dospělých pacientů s HER-2 negativním lokálně pokročilým inoperabilním nebo metastazujícím adenokarcinomem žaludku nebo gastroezofageální junkce, jejichž nádory exprimují PD-L1 se skóre pozitivity nádorové oblasti (TAP) nad 5%; v kombinaci s chemoterapií založenou na platině k léčbě první linie u dospělých pacientů s inoperabilním, lokálně pokročilým nebo metastazujícím spinocelulárním karcinomem jícnu, jejichž nádory exprimují PD-L1 se skóre TAP nad 5%.
Tivdak 40 mg prášek pro koncentrát pro infuzní roztok (tizotumab vedotin) – cytostatikum, konjugát protilátka-léčivo – monoterapie dospělých pacientek s recidivujícím nebo metastazujícím karcinomem děložního hrdla s progresí onemocnění během systémové terapie nebo po ní.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
12.4.2025
Aktualizace DUBEN 2025
Aktualizace AISLP duben 2025 je připravena ke stažení.
Vážení uživatelé programu AISLP,
rádi bychom Vám za celý náš tým popřáli krásné prožití velikonočních svátků.
Přinášíme Vám informace ke zveřejněným Spotřebám léčiv za rok 2024.
Pravidelně jednou ročně zveřejňujeme data ke spotřebě léčiv za rok 2024 - vycházíme z roční spotřeby léků podle openDat ze SÚKL (dodávky). Každoročně je doplňujeme údaji ze zpracovaných databází vystavených na stránkách www.nzip.cz - zde bývají vystaveny kvartální sumy spotřeb přes ATC ale také i pro jednotlivé léky i se souhrnem úhrad v korunách. Letos ještě stále na stránkách www.nzip.cz chybí tato data pro celý poslední kvartál roku 2024. Tyto údaje doplňujeme pro hrazené léky i součtem z openDat nazvaných LEK13. Letos nám ovšem nesedí celkový počet vydaných balení podle openDat dodavky oproti roční sumě LEK13 u asi poloviny léků (i pokud povolíme 15ti procentní odchylku mezi počty vydaných balení). Finanční sumy úhrad jsou proto doplněny jen u některých hrazených léků a celkové sumy úhrad pro celé ATC skupiny zatím neuvádíme.
Jakmile budou k dispozici další, Spotřeby postupně doplníme.
Následuje výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální dubnové verzi AISLP.
Avtozma 20 mg/ml koncentrát pro infuzní roztok a 162 mg injekční roztok v předplněné injekční stříkačce/peru (tocilizumab) – biosimilar, imunosupresivum, inhibitor interleukinu.
Fasenra 30 mg injekční roztok v předplněné injekční stříkačce
Fasenra 30 mg injekční roztok v předplněném peru (benralizumab) – rozšíření indikace, nově: přídatná léčba u dospělých pacientů s relabující nebo refrakterní eozinofilní granulomatózou s polyangiitidou (EGPA).
Ofev 25 mg, 100 mg, 150 mg měkké tobolky (nintedanib-esilát) – cytostatikum, inhibitor proteinkináz – rozšíření indikací, nově: léčba dětí a dospívajících od 6 do 17 let s klinicky významnými progredujícími fibrotizujícími intersticiálními plicními procesy (ILD); léčba, dospívajících a dětí od 6 let se systémovou sklerodermií s přidruženým intersticiálním plicním onemocněním (SSc-ILD).
Opdivo 10 mg/ml koncentrát pro infuzní roztok (nivolumab) – cytostatikum – rozšíření indikací, nově: v kombinaci s ipilimumabem k léčbě v první linii u dospělých pacientů s neresekovatelným nebo pokročilým hepatocelulárním karcinomem.
Paxneury 1 mg, 2 mg, 3 mg, 4 mg, 5 mg, 6 mg a 7 mg tablety s prodlouženým uvolňováním (guanfacin-hydrochlorid) – agonista centrálních alfa2-adrenoreceptorů – k léčbě poruchy pozornosti s hyperaktivitou (Attention Deficit Hyperactivity Disorder, ADHD) u dětí a dospívajících ve věku 6-17 let, pro které není vhodná léčba stimulancii, netolerují ji nebo se ukázala jako neúčinná. Přípravek se musí používat jakou součást komplexního léčebného programu ADHD, obvykle zahrnujícího psychologická, edukační a sociální opatření.
Plyzari 6 mg/ml injekční roztok v předplněném peru (liraglutid) – antidibetikum, GLP-1 agonista – první generikum s léčivou látkou liraglutid jako doplňková léčba k dietě se sníženým obsahem kalorií a zvýšené fyzické aktivitě za účelem úpravy hmotnosti u dospělých pacientů s počáteční hodnotou indexu tělesné hmotnosti (BMI) >=30 kg/m2 (obezita), nebo >=27 kg/m2 až <30 kg/m2 (nadváha) za přítomnosti alespoň jedné komorbidity související s tělesnou hmotností, např. s dysglykemií (prediabetes nebo diabetes mellitus 2. typu), hypertenzí, dyslipidemií nebo obstrukční spánkovou apnoí. Doplňková léčba ke zdravé výživě a zvýšené fyzické aktivitě k úpravě tělesné hmotnosti u dospívajících pacientů ve věku od 12 let s: obezitou (BMI odpovídající >=30 kg/m2 u dospělých podle mezinárodních hraničních hodnot), s tělesnou hmotností nad 60 kg.
Rekambys 600 mg a 900 mg injekční suspenze s prodlouženým uvolňováním (rilpivirin) – antivirotikum, nenukleosidový inhibitor reverzní transkriptázy – rozšíření indikace: přípravek je indikován v kombinaci s injekcemi kabotegraviru k léčbě infekce virem lidské imunodeficience typu 1 (HIV-1) u dospělých a dospívajících (od 12 let a s t.hm. min. 35 kg), kteří jsou virologicky suprimováni (RNA HIV-1 < 50 kopií/ml) na stabilním antiretrovirovém režimu bez důkazů současné nebo minulé virové rezistence na látky ze třídy nenukleosidových inhibitorů reverzní transkriptázy (NNRTI) a inhibitorů integrázy (INI) a bez virologického selhání při léčbě těmito látkami.
Rybrevant 350 mg koncentrát pro infuzní roztok (amivantamab) – cytostatikum, konjugáty protilátka-léčivo – rozšíření indikací, nově: v kombinaci s lazertinibem jako první linie léčby dospělých pacientů s pokročilým nemalobuněčným karcinomem plic (NSCLC) s delecemi v exonu 19 nebo substitučními mutacemi L858R v exonu 21 genu kódujícího EGFR.
Rytelo 47 mg, 188 mg prášek pro koncentrát pro infuzní roztok (imetelstat) – cytostatikum – monoterapie dospělých pacientů s anémií závislou na transfuzích v důsledku myelodysplastického syndromu (MDS) s velmi nízkým, nízkým nebo středním rizikem bez cytogenetické abnormality izolované delece 5q (non-del 5q), u nichž byla odpověď na léčbu erytropoetinem neuspokojivá nebo kteří nejsou pro léčbu na bázi erytropoetinu vhodní.
Tuzulby 20 mg, 30 mg a 40 mg žvýkací tablety s prodlouženým uvolňováním (methylfenidát) – nová léková forma methylfenidátu – součást komplexního léčebného programu pro poruchu pozornosti s hyperaktivitu (ADHD) u dětí a dospívajících ve věku od 6 do 17 let, kdy se samotná nápravná opatření ukáží jako nedostatečná. Léčba musí být pod dohledem specialisty na dětské poruchy chování. Diagnóza by měla být stanovena dle DSM-IV nebo pokynů v ICD-10 a měla by být založena na kompletní anamnéze a hodnocení pacienta. Diagnózu nelze stanovit pouze na základě přítomnosti jednoho nebo více příznaků.
Yervoy 5 mg/ml koncentrát pro infuzní roztok (ipilimumab) – cytostatikum, monoklonální protilátka – rozšíření indikací, nově: v kombinaci s nivolumabem k léčbě v první linii u dospělých pacientů s neresekovatelným nebo pokročilým hepatocelulárním karcinomem.
Yselty 100 mg, 200 mg potahované tablety (linzagolix-cholin) – gynekologikum – rozšíření indikace, nově: symptomatická léčba endometriózy u žen, které v minulosti podstoupily medikamentózní nebo chirurgickou léčbu endometriózy.
Vimkunya injekční suspenze v předplněné injekční stříkačce, vakcína proti onemocnění Chikungunya (rekombinantní, adsorbovaná) – k aktivní imunizaci k prevenci onemocnění způsobeného virem Chikungunya (CHIKV) u jedinců ve věku 12 let a starších. Vakcínu je třeba používat v souladu s oficiálními doporučeními.
Wainzua 45 mg injekční roztok v předplněném peru (eplontersen) – léčba polyneuropatie 1. nebo 2. stadia u dospělých pacientů s hereditární transthyretinovou amyloidózou (ATTRv).
Zefylti 30 MIU/0.5 ml a 48 MIU/0,5 ml injekční/infuzní roztok v předplněné injekční stříkačce (filgrastim) – biosimilar, cytokin, hemopoetický růstový faktor.
Zegluxen 6 mg/ml injekční roztok v předplněném peru (liraglutid) – antidibetikum, GLP-1 agonista – první generikum s léčivou látkou liraglutid k léčbě dospělých, dospívajících a dětí od 10 let s nedostatečně kontrolovaným diabetes mellitus 2. typu jako doplněk k dietě a cvičení: jako monoterapie, pokud je metformin považován za nevhodný v důsledku nesnášenlivosti nebo kontraindikací; jako přídavná léčba k dalším antidiabetikům.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
13.3.2025
Aktualizace BŘEZEN 2025
Aktualizace AISLP březen 2025 je připravena ke stažení.
Vážení uživatelé programu AISLP,
přinášíme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální březnové verzi AISLP.
Acarizax 12 SQ-HDM sublingvální lyofilizát, standardizovaný alergenový extrakt z roztočů domácího prachu Dermatophagoides pteronyssinus a Dermatophagoides farinae – přípravek pro alergenovou imunoterapii – rozšíření indikací: k léčbě pacientů s klinickou anamnézou a pozitivním testem citlivosti na roztoče domácího prachu (kožní prick test a/nebo specifické IgE) s nejméně jednou z následujících podmínek: středně těžká až těžká alergická rinitida u dospělých, dospívajících a dětí od 5 let způsobená roztoči domácího prachu přetrvávající i přes léčbu přípravky ulevujícími od příznaků; alergické astma u dospělých vyvolané roztoči domácího prachu, které není dobře kompenzované inhalačními kortikosteroidy a je spojené s mírnou až těžkou alergickou rinitidou způsobenou roztoči domácího prachu.
Andembry 200 mg injekční roztok v předplněné injekční stříkačce
Andembry 200 mg injekční roztok v předplněném peru (garadacimab) – rutinní prevence rekurentních atak hereditárního angioedému (HAE) u pacientů od 12 let.
Beyonttra 356 mg potahované tablety (akoramidis) – kardiakum – k léčbě transthyretinové amyloidózy divokého typu nebo variantní transthyretinové amyloidózy u dospělých pacientů s kardiomyopatií (ATTR-CM).
Bridion 100 mg/ml injekční roztok (sugammadex) – selektivní antagonista steroidních myorelaxancií – rozšíření věkového rozmezí u pediatrické indikace, nově: u dětí a dospívajících ve věku od narození do 17 let je sugammadex doporučen pouze pro běžné zrušení blokády navozené rokuroniem.
Emcitate 350 mikrogramů dispergovatelné tablety (tiratrikol) – hormon štítné žlázy – léčba periferní tyreotoxikózy u pacientů s deficitem monokarboxylátového transportéru 8 (MCT8) (Allanův-Herndonův-Dudleyův syndrom) od narození.
Hetronifly 10 mg/ml koncentrát pro infuzní roztok (serplulimab) – cytostatikum, monoklonální protilátka – v kombinaci s karboplatinou a etoposidem k první linii léčby dospělých pacientů s malobuněčným karcinomem plic v extenzivním stádiu (ES-SCLC).
Kavigale 300 mg injekční/infuzní roztok (sipavibart) – antivirová monoklonální protilátka – k preexpoziční profylaxi onemocnění covid-19 u dospělých a dospívajících pacientů ve věku 12 let a starších s t.hm. alespoň 40 kg, kteří jsou imunokompromitovaní vzhledem k zdravotnímu stavu nebo užívání imunosupresivní léčby. Přípravek má být používán v souladu s oficiálními doporučeními, pokud jsou k dispozici, a na základě informací o účinnosti sipavibartu proti v současnosti cirkulujícím virovým variantám.
Korjuny 10 mikrogramů a 50 mikrogramů koncentrát pro infuzní roztok (katumaxomab) – cytostatikum, monoklonální protilátka – intraperitoneální léčba maligního ascitu u dospělých s karcinomy pozitivními na epiteliální buněčné adhezní molekuly (EpCAM), kteří nejsou způsobilí k další systémové protinádorové léčbě.
Kostaive prášek pro injekční disperzi, vakcína sa-mRNA proti onemocnění covid-19 (zapomeran) – k aktivní imunizaci pro prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2 u osob ve věku 18 let a starších. Použití vakcíny má být v souladu s oficiálními doporučeními.
Lazcluze 80 mg a 240 mg potahované tablety (monohydrát lazertinib-mesilátu) – cytostatikum, inhibitor proteinkináz – v kombinaci s amivantamabem jako první linie léčby dospělých pacientů s pokročilým nemalobuněčným karcinomem plic (NSCLC) s delecemi v exonu 19 nebo substitučními mutacemi L858R v exonu 21 genu kódujícího EGFR.
Nemluvio 30 mg prášek a rozpouštědlo pro injekční roztok v předplněném peru/předplněné injekční stříkačce (nemolizumab) – dermatologikum, monoklonální protilátka – k léčbě středně těžké až těžké atopické dermatitidy (AD) u pacientů ve věku 12 let a starších, kteří jsou vhodnými kandidáty na systémovou léčbu. K léčbě dospělých se středně těžkým až těžkým prurigo nodularis (PN), kteří jsou vhodnými kandidáty na systémovou léčbu.
Nilemdo 180 mg potahované tablety (kyselina bempedoová) – hypolipidemikum – rozšíření indikací: nově k léčbě dospělých s prokázaným aterosklerotickým kardiovaskulárním onemocněním nebo s vysokým rizikem aterosklerotického kardiovaskulárního onemocnění za účelem snížení kardiovaskulárního rizika pomocí snížení hladiny LDL-C, jako doplněk ke korekci ostatních rizikových faktorů: u pacientů užívajících maximální tolerovanou dávkou statinu s ezetimibem nebo bez ezetimibu, nebo samostatně nebo v kombinaci s ezetimibem u pacientů, kteří statin netolerují nebo je u nich kontraindikován.
Nustendi 180 mg/10 mg potahované tablety (kyselina bempedoová/ezetimib) – hypolipidemikum – rozšíření indikací: nově k léčbě dospělých s prokázaným aterosklerotickým kardiovaskulárním onemocněním nebo s vysokým rizikem aterosklerotického kardiovaskulárního onemocnění za účelem snížení kardiovaskulárního rizika pomocí snížení hladiny LDL-C, jako doplněk ke korekci ostatních rizikových faktorů: u pacientů užívajících maximální tolerovanou dávku statinu, jejichž stav není dostatečně kontrolován doplňkovou léčbou ezetimibem, nebo u pacientů, kteří buď statin netolerují, nebo je u nich statin kontraindikován, a jejich stav není dostatečně kontrolován léčbou ezetimibem, nebo u pacientů již léčených kombinací kyseliny bempedoové a ezetimibu ve formě samostatných tablet.
Omvoh 300 mg koncentrát pro infuzní roztok a 100 mg a 200 mg injekční roztok v předplněné injekční stříkačce/peru (mirikizumab) – imunosupresivum, inhibitor interleukinu – nová síla přípravku 200 mg a rozšíření indikací, nově: léčba dospělých pacientů se středně těžkou až těžkou aktivní Crohnovou chorobou, u nichž byla odpověď na konvenční nebo biologickou léčbu nedostatečná, nebo u kterých došlo ke ztrátě odpovědi nebo tuto léčbu netolerovali.
Pyzchiva 45 mg a 90 mg injekční roztok v předplněném peru (ustekinumab) – biosimilar, imunosupresivum, inhibitor interleukinu – nová léková forma přípravku: k léčbě středně těžké až těžké plakové psoriázy u dospělých, u kterých selhaly jiné systémové léčby, včetně podávání cyklosporinu, methotrexátu (MTX) a PUVA (psoralen a ultrafialové záření A), nebo kteří tyto léčby netolerují nebo jsou u nich kontraindikovány. Samostatně nebo v kombinaci s MTX k léčbě aktivní psoriatické artritidy u dospělých pacientů, pokud odpověď na předchozí léčbu nebiologickými chorobu modifikujícími antirevmatickými léky (DMARD) nebyla dostatečná. K léčbě dospělých pacientů se středně těžkou až těžkou aktivní Crohnovou chorobou, u kterých buď odpověď na konvenční terapii nebo na antagonistu tumor nekrotizujícího faktoru alfa (TNF-alfa) nebyla dostatečná nebo odezněla nebo tito pacienti netolerovali konvenční léčbu nebo léčbu TNF-alfa, případně jsou u nich tyto terapie kontraindikovány.
Seladelpar Gilead 10 mg tvrdé tobolky (dihydrát seladelpar-lysinu) – léčivo k terapii onemocnění žlučových cest – léčba primární biliární cholangitidy (PBC) v kombinaci s kyselinou ursodeoxycholovou (UDCA) u dospělých pacientů, kteří mají nedostatečnou odpověď na samotnou UDCA, nebo jako monoterapie u dospělých pacientů s intolerancí UDCA.
Sirturo 20 mg a 100 mg tablety (bedachilin) – antituberkulotikum – rozšíření indikace: jako součást vhodné kombinované léčby u dospělých a pediatrických pacientů (od 5 let s t.hm. nejméně 15 kg), s plicní tuberkulózou (TB) vyvolanou bakterií Mycobacterium tuberculosis rezistentní přinejmenším na rifampicin a isoniazid. Je nutné respektovat oficiální pokyny pro správné používání antituberkulotik.
Vamipino 80 mg/1,5 mg tablety s řízeným uvolňováním (valsartan, indapamid)
Valsartan/Indapamid Krka 80 mg/1,5 tablety s řízeným uvolňováním – nová síla přípravku k léčbě esenciální hypertenze jako náhrada terapie u dospělých pacientů adekvátně kontrolovaných valsartanem a indapamidem podávanými současně ve stejné dávce jako v kombinaci, ale jako samostatné tablety.
Vocabria 400 mg a 600 mg injekční suspenze s prodlouženým uvolňováním (kabotegravir) – antivirotikum, inhibitor integrázy – rozšíření indikace: injekce přípravku Vocabria jsou v kombinaci s injekcemi rilpivirinu indikovány k léčbě infekce virem lidské imunodeficience typu 1 (HIV-1) u dospělých a dospívajících (od 12 let a s t.hm. min. 35 kg), u nichž bylo dosaženo virové suprese (HIV-1 RNA < 50 kopií/ml) na stabilním antiretrovirovém režimu bez průkazu stávající či předchozí virové rezistence na léčiva třídy nenukleosidových inhibitorů reverzní transkriptázy (NNRTI) a inhibitorů integrázy (INI) a u nichž v minulosti nedošlo při léčbě těmito léčivy k virologickému selhání.
Vocabria 30 mg potahované tablty (kabotegravir) – antivirotikum, inhibitor integrázy – rozšíření indikace: tablety přípravku Vocabria jsou v kombinaci s tabletami rilpivirinu indikovány ke krátkodobé léčbě infekce virem lidské imunodeficience typu 1 (HIV-1) u dospělých a dospívajících (od 12 let a s t.hm. min. 35 kg), u nichž bylo dosaženo virové suprese (HIV-1 RNA < 50 kopií/ml) na stabilním antiretrovirovém režimu bez průkazu stávající či předchozí virové rezistence na léčiva třídy nenukleosidových inhibitorů reverzní transkriptázy (NNRTI) a inhibitorů integrázy (INI) a u nichž v minulosti nedošlo při léčbě těmito léčivy k virologickému selhání, za následujícím účelem: perorální úvodní léčba k posouzení snášenlivosti kabotegraviru a rilpivirinu před podáním dlouhodobě působící injekce kabotegraviru + dlouhodobě působící injekce rilpivirinu; perorální léčba dospělých a dospívajících, kteří vynechají plánovanou dávku injekce kabotegraviru + injekce rilpivirinu.
Welireg 40 mg potahované tablety (belzutifan) – cytostatikum – monoterapie dospělých pacientů s pokročilým světlobuněčným karcinomem ledvin (RCC) a dospělých pacientů s von Hippelovou-Lindauovou chorobou (VHL), kteří vyžadují léčbu souvisejícího lokalizovaného karcinomu ledvin (RCC), hemangioblastomů centrálního nervového systému (CNS-HB) nebo neuroendokrinních nádorů pankreatu (pNET).
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
12.2.2025
Aktualizace ÚNOR 2025
Aktualizace AISLP únor 2025 je připravena ke stažení.
Vážení uživatelé programu AISLP,
zasíláme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální únorové verzi AISLP.
Augtyro 40 mg a 160 mg tvrdé tobolky (repotrektinib) – cytostatikum, inhibitor proteinkináz – monoterapie dospělých pacientů s ROS1-pozitivním pokročilým nemalobuněčným karcinomem plic (NSCLC); monoterapie dospělých a pediatrických pacientů od 12 let s pokročilými solidními nádory s fúzí genu NTRK, kteří byli dříve léčeni inhibitorem NTRK nebo dříve nebyli léčeni inhibitorem NTRK a léčebné možnosti necílící na NTRK mají omezený klinický přínos nebo byly vyčerpány.
Bimervax XBB.1.16 injekční emulze, vakcína proti onemocnění covid-19, rekombinantní, obsahující adjuvans (damlekovatein) – k aktivní imunizaci k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2 u osob ve věku 16 let a starších. Použití vakcíny má být v souladu s oficiálními doporučeními.
Bravilon 50 mg mukoadhezivní bukální tablety (aciklovir) – k léčbě rekurentního onemocnění herpes labialis u imunokompetentních dospělých s častými epizodami exacerbace infekce způsobené herpes simplex virem.
Evkeeza 150 mg/ml koncentrát pro infuzní roztok (evinakumab) – hypolipidemikum, monoklonální protilátka – rozšíření indikace: přídavný prostředek k dietě a jiným terapiím ke snížení LDL-cholesterolu (LDL-C) k léčbě dospělých a pediatrických pacientů ve věku 6 měsíců a starších s homozygotní familiární hypercholesterolemií (HoFH).
Flucelvax Tetra injekční suspenze v předplněné injekční stříkačce, vakcína proti chřipce (povrchový antigen, inaktivovaná, připravená na buněčných kulturách) – rozšíření indikace: profylaxe chřipky u dospělých a dětí od 6 měsíců věku. Přípravek má být podáván v souladu s oficiálními doporučeními.
Gohibic 200 mg koncentrát pro infuzní roztok (vilobelimab) – léčba dospělých pacientů se syndromem akutní respirační tísně (ARDS) vyvolaným virem SARS-CoV2, kterým jsou v rámci standardní léčby podávány systémové kortikosteroidy a kteří jsou připojeni na invazivní mechanickou ventilaci (IMV) (s mimotělní membránovou oxygenací (ECMO) nebo bez ní).
Hukyndra 20 mg injekční roztok v předplněné injekční stříkačce (adalimumab) – nová síla přípravku, pro pediatrické pacienty – v kombinaci s methotrexátem k léčbě aktivní polyartikulární juvenilní idiopatické artritidy u pacientů od 2 let, u kterých odpověď na léčbu jedním nebo více chorobu modifikujícími antirevmatiky (DMARDs) nebyla dostatečná; při nesnášenlivosti methotrexátu nebo v případě, kdy pokračování v léčbě methotrexátem není vhodné, může být přípravek Hukyndra podáván samostatně. Entezopatická artritida: léčba aktivní entezopatické artritidy u pacientů od 6 let, u nichž nebylo dosaženo adekvátní odpovědi na konvenční léčbu, nebo u nichž léčba nebyla tolerována. Léčba těžké chronické ložiskové psoriázy u dětí a dospívajících od 4 let, u kterých reakce na lokální terapii a fototerapie nebyla dostatečná nebo nejsou pro tuto léčbu vhodnými kandidáty. Léčba středně těžké až těžké aktivní Crohnovy choroby u pediatrických pacientů (od 6 let), u kterých reakce na konvenční léčbu, včetně primární nutriční léčby a kortikosteroidů a/nebo imunosupresiv nebyla dostatečná nebo kteří ji netolerují nebo je u nich tato léčba kontraindikována. Léčba chronické neinfekční přední uveitidy u pediatrických pacientů ve věku od 2 let, u kterých reakce na konvenční léčbu nebyla dostatečná nebo kteří ji netolerují nebo u nichž tato léčba není vhodná.
Jakavi 5 mg/ml perorální roztok (ruxolitinib-fosfát) – cytostatikum, inhibitor proteinkináz – nová léková forma přípravku: k léčbě dospělých a pediatrických pacientů od 28 dnů s akutní reakcí štěpu proti hostiteli, kteří adekvátně nereagují na léčbu kortikosteroidy nebo jinou systémovou léčbu; k léčbě dospělých a pediatrických pacientů od 6 měsíců s chronickou reakcí štěpu proti hostiteli, kteří adekvátně nereagují na léčbu kortikosteroidy nebo jinou systémovou léčbu.
Kevzara 175 mg/ml injekční roztok (sarilumab) – nová léková forma přípravku pro pediatrické pacienty – léčba aktivní polyartikulární juvenilní idiopatické artritidy (pJIA; polyartritida s pozitivním nebo negativním revmatoidním faktorem a extendovaná oligoartritida) u pacientů ve věku od 2 let, kteří neodpovídají adekvátně na předchozí léčbu konvenčními syntetickými chorobu modifikujícími antirevmatickými léky (csDMARD), v monoterapii nebo v kombinaci s methotrexátem (MTX).
Kisqali 200 mg potahované tablety (ribociklib-sukcinát) – cytostatikum, inhibitor proteinkináz – rozšíření indikací: nově v kombinaci s inhibitorem aromatázy k adjuvantní léčbě pacientů s časným karcinomem prsu s pozitivitou hormonálních (HR) receptorů a negativitou receptorů 2 pro lidský epidermální růstový faktor (HER-2) s vysokým rizikem recidivy.
Nutriflex Omega special bez elektrolytů infuzní emulze – rozšíření indikace pro děti od 2 let.
Nutriflex Omega special 56/144 infuzní emulze – rozšíření indikace pro děti od 2 let.
Nutriflex Omega Peri infuzní emulze – rozšíření indikace pro děti od 2 let.
Palforzia 0,5 mg, 1 mg, 10 mg, 20 mg a 100 mg perorální prášek v tobolkách pro otevření a 300 mg perorální prášek v sáčku (odtučněný podzemnicový prášek) – přípravek pro alergenovou imunoterapii – rozšíření indikace: k léčbě pacientů ve věku 1 až 17 let s potvrzenou diagnózou alergie na burské oříšky. U pacientů od 18 let se může v užívání přípravku pokračovat. Přípravek se má používat ve spojení s dietou s vyloučením burských oříšků.
Pegasys 90 mikrogramů, 135 mikrogramů a 180 mikrogramů injekční roztok v předplněné injekční stříkačce (peginterferon alfa-2a) – nové indikace: monoterapie onemocnění polycythaemia vera u dospělých; monoterapie esenciální trombocytemie u dospělých.
SIILTIBCY (0,5 mikrogramu + 0,5 mikrogramu)/ml injekční roztok (rdESAT-6, rCFP-10) – diagnostikum – detekce infekce Mycobacterium tuberculosis, včetně onemocnění, u dospělých a dětí ve věku 28 dní a starších.
Synjardy 5 mg/850 mg, 5 mg/1000 mg, 12,5 mg/850 mg a 12,5 mg/1000 mg potahované tablety (empagliflozin, metformin-hydrochlorid) – rozšíření indikace: léčba diabetes mellitus 2. typu u dospělých a dětí ve věku 10 let a starších jako doplněk diety a tělesného cvičení: u pacientů nedostatečně kontrolovaných na maximální tolerované dávce metforminu podávaného v monoterapii; v kombinaci s jinými léčivými přípravky k léčbě diabetu u pacientů nedostatečně kontrolovaných metforminem a těmito léčivými přípravky; u pacientů, kteří jsou již léčeni kombinací empagliflozinu a metforminu v samostatných tabletách.
Uzpruvo 130 mg koncentrát pro infuzní roztok (ustekinumab) – biosimilar.
Vamipino 80 mg/1,5 mg tablety s řízeným uvolňováním (valsartan, indapamid) – nová síla přípravku k léčbě esenciální hypertenze jako náhrada terapie u dospělých pacientů adekvátně kontrolovaných valsartanem a indapamidem podávanými současně ve stejné dávce jako v kombinaci, ale jako samostatné tablety.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
31.1.2025
Novinky roku 2024 v AISLP
Podívejte se s námi, co jsme pro Vás v AISLP během roku 2024 připravili.
Pokud se Vám video nezobrazuje, přejděte prosím na náš účet na Youtube - AISLP Developer.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
14.1.2025
Aktualizace LEDEN 2025
Aktualizace AISLP leden 2025 je připravena ke stažení.
Vážení uživatelé programu AISLP,
zasíláme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální lednové verzi AISLP.
Zároveň v aktuální verzi programu AISLP naleznete v našem seznamu nové ATC skupiny a jejich změny platné pro rok 2025, jak je uvádí WHO Collaborating Centre for Drug Statistics Methodology. Zařazení léčivých látek a léčivých přípravků je již v souladu s ATC/DDD Indexem 2025.
Absimky 130 mg koncentrát pro infuzní roztok a 45 mg a 90 mg injekční roztok v předplněné injekční stříkačce (ustekinumab) – biosimilar.
Aflunov injekční suspenze v předplněné injekční stříkačce, vakcína proti zoonotické chřipce (H5N1), (povrchový antigen, inaktivovaná, s adjuvans) – rozšíření indikace: aktivní imunizace proti subtypu H5N1 chřipkového viru A u jedinců od 6 měsíců věku. Vakcína se má používat v souladu s oficiálním doporučením.
Alhemo 15 mg/1,5 ml, 60 mg/1,5 ml, 150 mg/1,5 ml a 300 mg/3 ml injekční roztok v předplněném peru (koncizumab) – rutinní profylaxe krvácení u pacientů od 12 let s hemofilií A s inhibitory FVIII nebo s hemofilií B s inhibitory FIX.
Cerdelga nová síla 21 mg tvrdé tobolky (eliglustat-tartarát) – trávicí trakt a metabolismus – rozšíření indikace, nově: léčba dětí od 6 do 18 let s tělesnou hmotností od 15 kg s Gaucherovou chorobou typu 1 (GD1), které jsou stabilizované na enzymatické substituční terapii (ERT) a jsou pomalými (PMs), středně rychlými (IMs) nebo rychlými (EMs) metabolizátory substrátů enzymu CYP2D6.
Dupixent 200 mg a 300 mg injekční roztok v předplněné injekční stříkačce/předplněném peru (dupilumab) – rozšíření indikací, nově: k léčbě eozinofilní ezofagitidy u dospělých, dospívajících a dětí od 1 roku s t.hm. alespoň 15 kg, jejichž onemocnění není dostatečně kontrolováno konvenční léčbou, netolerují ji nebo, kteří nejsou pro konvenční léčbu vhodnými kandidáty.
Esperoct 500 IU, 1000 IU, 1500 IU, 2000 IU, 3000 IU, 4000 IU a 5000 IU prášek a rozpouštědlo pro injekční roztok (turoktokog alfa pegol) – rozšíření indikace na všechny věkové skupiny.
Everolimus Teva 5 mg, 10 mg tablety (everolimus) – cytostatikum, inhibitor mTOR – rozšíření indikací, nově: léčba dospělých pacientů s renálním angiomyolipomem spojeným s komplexem tuberózní sklerózy (TSC); léčba dospělých a pediatrických pacientů se subependymálním obrovskobuněčným astrocytomem (SEGA) spojeným s TSC.
Hepcludex 2 mg prášek pro injekční roztok (bulevirtid) – antivirotikum – rozšíření indikace: k léčbě chronické infekce virem hepatitidy delta (HDV) u HDV-RNA pozitivních (v plazmě nebo séru) dospělých a pediatrických pacientů ve věku 3 let a starších s t.hm. minimálně 10 kg s kompenzovaným onemocněním jater.
Imuldosa 130 mg koncentrát pro infuzní roztok a 45 mg a 90 mg injekční roztok v předplněné injekční stříkačce (ustekinumab) – biosimilar.
Leponex 200 mg tablety (klozapin) – nová síla přípravku – léčba rezistentní formy schizofrenie u dospělých pacientů, kteří mají závažné neléčitelné neurologické nežádoucí účinky na podávání ostatních antipsychotik, včetně atypických. Rezistence na léčbu je definována jako nedostatečné klinické zlepšení navzdory podávání nejméně dvou rozdílných antipsychotik, včetně atypických, v dostatečných dávkách po odpovídající dobu. Léčba psychotických poruch u dospělých pacientů s Parkinsonovou chorobou, u kterých selhala standardní terapie.
Lynparza 100 mg, 150 mg potahované tablety (olaparib) – cytostatikum – rozšíření indikací, nově: v kombinaci s durvalumabem k udržovací léčbě dospělých pacientek s primárně pokročilým nebo rekurentním karcinomem endometria, který je mismatch repair proficientní (pMMR), pokud onemocnění neprogredovalo při léčbě durvalumabem v kombinaci s karboplatinou a paklitaxelem v první linii léčby.
Midazolam AVMC 7,5 mg a 15 mg potahované tablety (midazolam) – krátkodobá léčba nespavosti; pouze v případě, že porucha je závažná, zneschopňující nebo způsobuje jedinci výraznou nepohodu. Sedace při premedikaci před chirurgickými nebo diagnostickými výkony. Pro dospělé a dospívající od 12 let.
Nesyrgy 5 mg/5 mg a 5 mg/10 mg potahované tablety (nebivolol, amlodipin) – nová fixní kombinace k léčbě esenciální hypertenze, jako náhrada terapie u dospělých pacientů adekvátně kontrolovaných nebivololem a amlodipinem podávanými současně ve stejné dávce jako v kombinaci, ale jako samostatné tablety.
Nutriflex Omega plus 38/120 infuzní emulze – rozšíření indikace pro děti od 2 let.
Tobramycin Vioser 1 mg/ml infuzní roztok (tobramycin) – aminoglykosidové antibiotikum – k léčbě závažných infekcí způsobených patogeny citlivými na tobramycin u dospělých, dospívajících a dětí ve věku od 1 týdne. Hlavními indikacemi jsou infekce způsobené patogeny rezistentními k jiným, méně toxickým lékům, závažné infekce způsobené gramnegativními patogeny, nozokomiální infekce a infekce u imunokompromitovaných pacientů a pacientů s neutropenií. Přípravek lze použít k léčbě: komplikovaných infekcí močových cest, infekcí dolních cest dýchacích, včetně nozokomiální pneumonie, intraabdominálních infekcí, endokarditidy, meningitidy způsobené gramnegativními patogeny, osteomyelitidy a purulentní artritidy, bakteriemie související s výše uvedenými infekcemi nebo při podezření na tuto souvislost. Je třeba vzít v úvahu oficiální doporučení pro správné použití antibakteriálních látek.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
13.12.2024
Aktualizace PROSINEC 2024
Aktualizace AISLP prosinec 2024 je připravena ke stažení.
Vážení uživatelé programu AISLP,
zasíláme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální prosincové verzi AISLP.
Buccolam 2,5 mg, 5 mg, 7,5 mg a 10 mg orální roztok (midazolam) – rozšíření indikací na dospělé pacienty – léčba prolongovaných akutních záchvatů křečí u dětí od 3 měsíců a dospělých.
Elahere 5 mg/ml koncentrát pro infuzní roztok (mirvetuximab soravtansin) – cytostatikum, monoklonální protilátka – monoterapie dospělých pacientek s high grade serózním epiteliálním karcinomem ovaria, tubárním karcinomem nebo primárním karcinomem peritonea s pozitivitou folátového receptoru alfa (FRalfa) a rezistencí k léčbě na bázi platiny, které podstoupily jeden až tři předchozí režimy systémové léčby.
Hympavzi 150 mg injekční roztok v předplněné injekční stříkačce.
Hympavzi 150 mg injekční roztok v předplněném peru (marstacimab) – rutinní profylaxe krvácivých příhod u pacientů od 12 let, vážících alespoň 35 kg, s těžkou hemofilií A (vrozený nedostatek faktoru VIII, FVIII <1%) bez inhibitorů faktoru VIII nebo s těžkou hemofilií B (vrozený nedostatek faktoru IX, FIX <1%) bez inhibitorů faktoru IX.
Orfiril long 150 mg a 300 mg tvrdé tobolky s prodlouženým uvolňováním a 500 mg a 1000 mg granule s prodlouženým uvolňováním v sáčku (kyselina valproová) – zrušení indikace: profylaxe migrény u dospělých a dospívajících od 12 let.
Ozawade 4,5 mg a 18 mg potahované tablety (pitolisant) – zlepšení bdělosti a snížení nadměrné denní spavosti (excessive daytime sleepiness, EDS) u dospělých pacientů s obstrukční spánkovou apnoe (obstructive sleep apnoea, OSA), jejichž EDS není úspěšně léčena primární terapií OSA, jako je kontinuální přetlak v dýchacích cestách (continuous positive airway pressure, CPAP), nebo kteří tuto léčbu netolerují. Aktualizace SmPC – zvýšení maximální denní dávky na 36 mg.
Penbraya prášek a suspenze pro injekční suspenzi, konjugovaná vakcína proti meningokokům skupiny A, C, W, Y a vakcína proti meningokokům skupiny B (rekombinantní, adsorbovaná) – k aktivní imunizaci jedinců starších 10 let k zabránění invazivnímu onemocnění způsobenému Neisseria meningitidis skupiny A, B, C, W a Y. Vakcínu je třeba používat v souladu s oficiálními doporučeními.
Qsiva 3,75 mg/23 mg, 7,5 mg/46 mg, 11,25 mg/69 mg, 15 mg/92 mg tvrdé tobolky s řízeným uvolňováním (fentermin-hydrochlorid, topiramát) – léčivo k terapii obezity – doplněk k nízkokalorické dietě a k tělesné aktivitě, k regulaci tělesné hmotnosti u dospělých pacientů s počátečním indexem tělesné hmotnosti (BMI): od 30 kg/m2 (obézní pacienti) nebo od 27 kg/m2 (pacienti s nadváhou) s komorbiditami souvisejícími s tělesnou hmotností, jako je hypertenze, diabetes 2. typu nebo dyslipidemie.
Tagrisso 40 mg, 80 mg potahované tablety (osimertinib) – cytostatikum, inhibitor proteinkináz – rozšíření indikace: nově také v kombinaci s pemetrexedem a chemoterapií na bázi platiny pro léčbu první linie dospělých pacientů s pokročilým NSCLC, jejichž nádory mají delece v exonu 19 EGFR nebo substituční mutaci v exonu 21 (L858R).
Tepkinly 4 mg/0,8 ml, 48 mg injekční roztok (epkoritamab) – cytostatikum, konjugát protilátka-léčivo – rozšíření indikace: nově také v monoterapii k léčbě dospělých pacientů s relabujícím nebo refrakterním folikulárním lymfomem (FL) po dvou nebo více liniích systémové léčby.
Zavicefta 2 g/0,5 g prášek pro koncentrát pro infuzní roztok (ceftazidim/avibaktam) – cefalosporinové antibiotikum III. generace s inhibitorem beta-laktamáz – rozšíření indikace: léčba následujících infekcí u dospělých a pediatrických pacientů od narození: komplikované intraabdominální infekce, komplikované infekce močových cest včetně pyelonefritidy, nozokomiální pneumonie včetně ventilátorové pneumonie. Léčba dospělých pacientů s bakteriemií, která se vyskytne v souvislosti s kteroukoli výše zmíněnou infekcí nebo u níž existuje podezření na tuto souvislost. Přípravek je též indikován k léčbě infekcí vyvolaných gramnegativními aerobními mikroorganismy u dospělých a pediatrických pacientů od narození s omezenými léčebnými možnostmi. Je třeba respektovat oficiální doporučení o vhodném používání antibakteriálních látek.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
2.12.2024
Nová adresa firmy INPHARMEX, spol. s r.o.
Od prosince 2024 nás najdete na nové adrese.
Vážení uživatelé AISLP,
rádi bychom Vám oznámili, že naše firma změnila korespondenční a fakturační adresu:
INPHARMEX, spol. s r.o.
Ohradské náměstí 1628/7
155 00 Praha 5, Stodůlky
Váš tým AISLP
www.aislp.cz
12.11.2024
Aktualizace LISTOPAD 2024
Aktualizace AISLP listopad 2024 je připravena ke stažení.
Vážení uživatelé programu AISLP,
zasíláme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální listopadové verzi AISLP.
Arexvy prášek a suspenze pro injekční suspenzi, vakcína proti respiračnímu syncytiálnímu viru (RSV) (rekombinantní, obsahující adjuvans) – rozšíření indikace: k aktivní imunizaci za účelem prevence onemocnění dolních cest dýchacích způsobeného respiračním syncytiálním virem u: dospělých ve věku 60 let a starších; dospělých ve věku 50 až 59 let se zvýšeným rizikem onemocnění RSV. Použití vakcíny má být v souladu oficiálními doporučeními.
Blossomin potahované tablety (suchý extrakt z natě mučenky) – tradiční rostlinný léčivý přípravek používaný k úlevě od mírných příznaků duševního napětí, jako jsou nervozita, obavy nebo podrážděnost, a k podpoře spánku. Použití je založeno výlučně na zkušenosti z dlouhodobého použití.
Edurant 25 mg potahované tablety (rilpivirin) – rozšíření indikace: v kombinaci s jinými antiretrovirovými léčivými přípravky k léčbě infekce virem lidské imunodeficience typu 1 (HIV-1) u dospělých a pediatrických pacientů s t.hm. nejméně 25 kg bez známých mutací spojených s rezistencí ke třídě nenukleosidových inhibitorů reverzní transkriptázy (NNRTI) a s virovou náloží <= 100 tis. HIV-1 RNA kopií/ml. Při užívání přípravku je nutno se řídit testováním genotypové rezistence.
Edurant 2,5 mg dispergovatelné tablety (rilpivirin) – nová síla a léková forma: v kombinaci s jinými antiretrovirovými léčivými přípravky k léčbě infekce virem lidské imunodeficience typu 1 (HIV-1) u pediatrických pacientů ve věku 2 až méně než 18 let s t.hm. nejméně 14 kg až méně než 25 kg bez známých mutací spojených s rezistencí ke třídě nenukleosidových inhibitorů reverzní transkriptázy (NNRTI) a s virovou náloží <= 100 tis. HIV-1 RNA kopií/ml. Při užívání přípravku je nutno se řídit testováním genotypové rezistence.
Fymskina 45 mg a 90 mg injekční roztok v předplněné injekční stříkačce, 130 mg koncentrát pro infuzní roztok (ustekinumab) – biosimilar.
Imvanex injekční suspenze, vakcína proti pravým neštovicím a opičím neštovicím (živý modifikovaný virus vakcínie Ankara) – rozšíření indikace: aktivní imunizace proti pravým neštovicím, opičím neštovicím a onemocnění způsobenému virem vakcínie u jedinců ve věku 12 let a starších. Použití vakcíny má být v souladu s oficiálními doporučeními.
Imcevree 10 mg/ml injekční roztok (setmelanotid) – léčivo k terapii obezity – rozšíření indikace: léčba obezity a kontrola hladu spojeného s geneticky potvrzeným Bardetovým-Biedlovým syndromem (BBS), bialelickým deficitem proopiomelanokortinu (POMC), včetně PCSK1, vedoucím ke ztrátě funkce, nebo bialelickým deficitem leptinového receptoru (LEPR) u dospělých a nově dětí od 2 let (dříve od 6 let).
Imfinzi 50 mg/ml koncentrát pro infuzní roztok (durvalumab) – cytostatikum, monoklonální protilátka – rozšíření indikace: nově v kombinaci s karboplatinou a paklitaxelem v první linii léčby dospělých pacientek s primárně pokročilým nebo rekurentním karcinomem endometria, které jsou kandidátkami na systémovou terapii, po níž následuje udržovací léčba: durvalumabem v monoterapii u karcinomu endometria, který je mismatch repair deficientní (dMMR); durvalumabem v kombinaci s olaparibem u karcinomu endometria, který je mismatch repair proficientní (pMMR).
Nuvaxovid JN.1 injekční disperze, vakcína proti onemocnění covid-19 (rekombinantní, obsahující adjuvans) – k aktivní imunizaci osob ve věku 12 let a starších k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2. Vakcínu je třeba používat v souladu s oficiálními doporučeními.
Opsumit 10 mg potahované tablety (macitentan) – rozšíření indikace: nově také k dlouhodobé léčbě plicní arteriální hypertenze (PAH) funkční třídy II až III dle klasifikace WHO u pediatrických pacientů s t.hm. >= 40 kg, v monoterapii nebo kombinované terapii.
Opsumit 2,5 mg dispergovatelné tablety tablety (macitentan) – nová síla a léková forma: k dlouhodobé léčbě plicní arteriální hypertenze (PAH) funkční třídy II až III dle klasifikace WHO u pediatrických pacientů ve věku 2 roky až méně než 18 let, v monoterapii nebo kombinované terapii.
Otezla 10 mg, 20 mg a 30 mg potahované tablety (apremilast) – rozšíření indikací na pediatrické pacienty, nová balení: léčba středně závažné až závažné chronické ložiskové psoriázy u dětí a dospívajících ve věku od 6 let s t.hm. alespoň 20 kg, u nichž připadá v úvahu systémová léčba.
Slenyto 1 mg a 5 mg tablety s prodlouženým uvolňováním (melatonin) – změna indikací: léčba insomnie u dětí a dospívajících ve věku 2-18 let s poruchou autistického spektra (autism spectrum disorder, ASD) a/nebo s neurogenetickými poruchami s aberantní 24hodinovou sekrecí melatoninu a/nebo nočním probouzením, kde nejsou opatření v oblasti hygieny spánku dostatečná.
Spevigo 450 mg koncentrát pro infuzní roztok (spesolimab) – rozšíření indikací na pediatrické pacienty: léčba vzplanutí generalizované pustulózní psoriázy (GPP) u dospělých a dospívajících ve věku od 12 let v monoterapii.
Spevigo 150 mg injekční roztok v předplněné injekční stříkačce (spesolimab) – nová léková forma: prevence vzplanutí generalizované pustulózní psoriázy (GPP) u dospělých a dospívajících ve věku od 12 let.
Tedenomo 40 mg/1,5 mg a 80 mg/1,5 mg tablety s řízeným uvolňováním (telmisartan, indapamid) – k léčbě esenciální hypertenze jako náhrada terapie u dospělých pacientů adekvátně kontrolovaných telmisartanem a indapamidem podávanými současně ve stejných dávkách jako v kombinaci, ale jako samostatné tablety.
Tevimbra 100 mg koncentrát pro infuzní roztok (tislelizumab) – cytostatikum, monoklonální protilátka – rozšíření indikací: nově k léčbě nemalobuněčného karcinomu plic (NSCLC) v kombinaci s chemoterapií obsahující pemetrexed a platinu, karboplatinu a paklitaxel nebo nab-paklitaxel nebo v monoterapii.
Triumeq 5 mg/60 mg/30 mg dispergovatelné tablety (dolutegravir, abakavir, lamivudin) – rozšíření indikace: k léčbě infekce virem lidské imunodeficience typu 1 (HIV-1) u dětí ve věku nejméně 3 měsíce s t.hm. alespoň 6 kg a méně než 25 kg. Před zahájením léčby přípravky obsahujícími abakavir je nutno u jakéhokoli pacienta infikovaného HIV, bez ohledu na jeho rasový původ, provést vyšetření na přítomnost alely HLA-B*5701. Abakavir nemají užívat pacienti, o nichž je známo, že jsou nositeli alely HLA-B*5701.
Vabysmo 120 mg/ml injekční roztok (faricimab) – oftalmologikum – rozšíření indikací: nově k léčbě dospělých pacientů s poruchou zraku způsobenou makulárním edémem při okluzi retinální žíly (RVO; okluze větve retinální žíly nebo okluze centrální retinální žíly).
Yuvanci 10 mg/20 mg a10 mg/40 mg potahované tablety (macitentan, tadalafil) – náhrada léčby pro dlouhodobou terapii plicní arteriální hypertenze (PAH) u dospělých pacientů s funkční třídou II až III dle WHO, kteří jsou již léčeni kombinací macitentanu a tadalafilu podávanými souběžně ve formě samostatných tablet.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
11.11.2024
XXVI. sympozium klinické farmacie René Macha
Letos se opět účastníme sympozia klinické farmacie René Macha v Mikulově.
Novinkou, kterou budeme na sympoziu představovat je funkcionalita "kontrola maximálních dávek léčivých přípravků", kterou společnost Inpharmex vyvinula ve spolupráci se společností STAPRO.

Budeme se na Vás těšit v Mikulově!
Váš tým AISLP
www.aislp.cz
25.10.2024
Společný projekt společností INPHARMEX a STAPRO
Kontrola maximálních dávek léčivých přípravků
Vážení uživatelé AISLP,
rádi bychom Vás informovali o společném projektu, na kterém se podílí firma INPHARMEX, Farmaceutická fakulta UK Hradec Králové a společnost STAPRO. Jedná se o funkcionalitu "Kontrola dávkování léků", která je již nyní součástí nemocničního informačního systému FONS Enterprise společnosti STAPRO (webového i klasického klienta). Medikační pochybení při pobytu v nemocnici představují vážné nebezpečí pro pacienty. Vyvinutá funkcionalita automaticky kontroluje dávky léčivých přípravků ordinovaných ve strukturované medikaci nebo v receptu dle hodnot maximální, doporučené nebo obvyklé dávky z SmPC daného léčivého přípravku s ohledem na základní kritéria pacienta.
Dávka se kontroluje jako součet všech dávek podávaných v medikačním dni (denní dávka), případně jako dávka jednotlivá. Maximální nebo doporučené dávky jsou stanoveny pro běžně užívané léčivé přípravky v nemocnicích ve fázi udržovací léčby. Dávky jsou zpracovány do požadovaného strukturovaného formátu a v online režimu zobrazovány prostřednictvím Api rozhraní do FONS Enterprise. Systém FONS Enterprise provádí vyhodnocení předaných informací a zobrazuje upozornění v případech, kdy ordinované dávky nejsou v souladu s doporučením nebo nemohou být vyhodnoceny.
Kontrola maximálních dávek léků pracuje s účinnými látkami, pokud se v medikaci pacienta objeví dvě generika s toutéž účinnou látkou, jsou vyhodnocena ve vzájemné souvislosti.
Barevná indikace výsledku Kontroly maximálních dávek léků je zobrazena v poli Rozpis nebo Dávka.
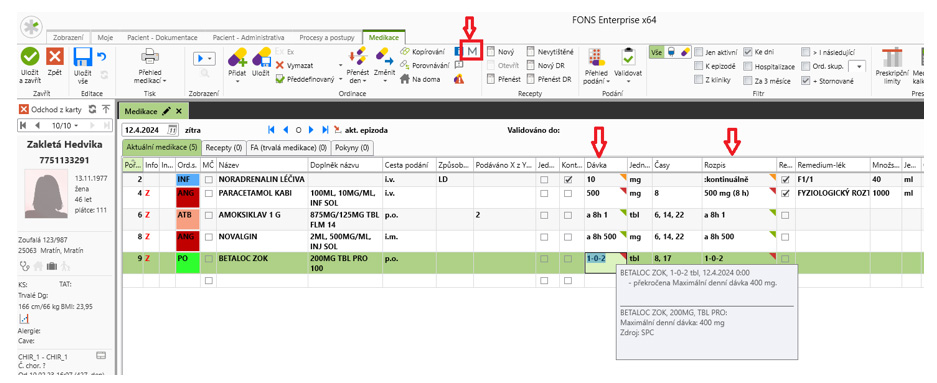
Rychlost a funkčnost celého řešení je nyní testována s možností využít bezplatného období. Již dnes je v systému zařazeno více než 30 nemocnic a systém zvládá online v reálném čase zodpovědět více než 100 000 dotazů týdně. K plnému využití všech možností, případně zobrazení kompletních informací o léčivém přípravku je podmínkou existence licence k programu AISLP.
V případě Vašeho zájmu dozvědět se více nebo se připojit k možnosti vyzkoušet si systém, kontaktujete buď přímo společnost STAPRO nebo naši společnost INPHARMEX.
Váš tým AISLP
www.aislp.cz
16.10.2024
Setkání uživatelů klinických modulů společnosti STAPRO
Ve dnech 16. - 17. října 2024 se budeme účastnit Setkání uživatelů klinických modulů společnosti STAPRO s. r. o.
Na setkání budeme prezentovat funkcionalitu "kontrola maximálních dávek", kterou společnost Inpharmex vyvinula ve spolupráci se společností Stapro.
Za náš tým se setkání zúčastní Vladimír Chmela, Alena Trojáčková a Martin Gurovič.
www.aislp.cz
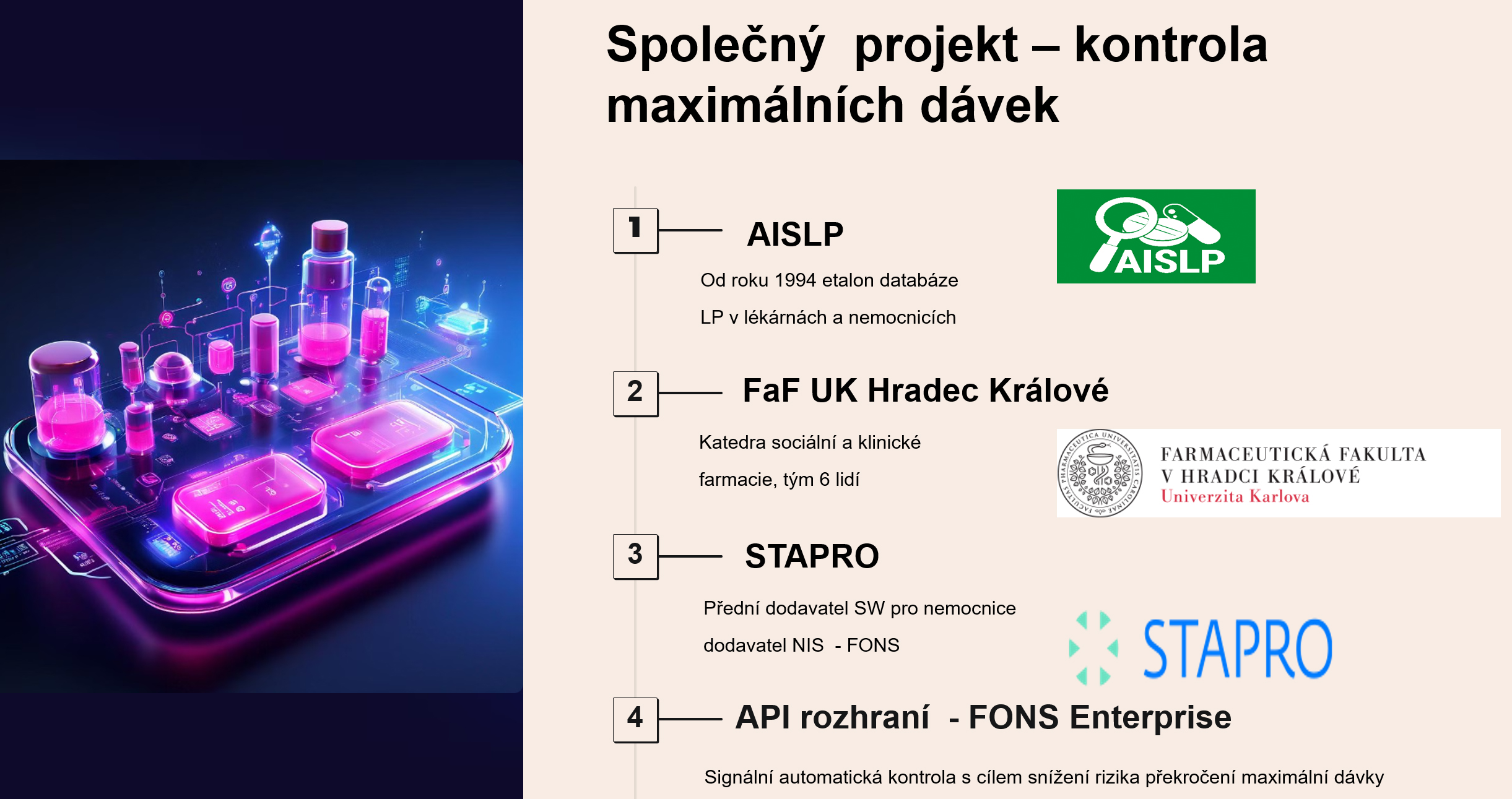
16.10.2024
SÚKL informuje o uvolnění distribuce a výdeje uvedených šarží léčivého přípravku Řepíková nať, spc. 50g.
16.10.2024
SÚKL informuje o stažení uvedené šarže léčivého přípravku Řepíková nať, spc. 50g až z úrovně zdravotnických zařízení.
14.10.2024
Aktualizace ŘÍJEN 2024
Aktualizace AISLP říjen 2024 je připravena ke stažení.
Vážení uživatelé programu AISLP,
zasíláme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální říjnové verzi AISLP.
Anzupgo 20 mg/g krém (delgocitinib) – dermatologikum, inhibitor Janus kináz – k léčbě středně těžkého až těžkého chronického ekzému rukou (chronic hand eczema, CHE) u dospělých, u kterých jsou lokální kortikosteroidy nedostatečně účinné nebo nejsou vhodné.
Betmiga 25 mg, 50 mg tablety s prodlouženým uvolňováním + nová léková forma 8 mg/ml granule pro perorální suspenzi s prodlouženým uvolňováním (mirabegron) – močové spazmolytikum – rozšíření indikace: nově léčba neurogenní hyperaktivity detruzoru (NDO) u pediatrických pacientů ve věku 3-18 let.
Beyfortus 50 mg a 100 mg injekční roztok v předplněné injekční stříkačce (nirsevimab) – antivirová monoklonální protilátka – rozšíření indikace: k prevenci onemocnění dolních cest dýchacích způsobeného respiračním syncytiálním virem (RSV) u: novorozenců a kojenců během jejich první sezóny RS; dětí do věku 24 měsíců, u nichž přetrvává riziko závažného onemocnění RSV během jejich druhé sezóny RSV. Přípravek má být používán v souladu s oficiálními doporučeními.
Braftovi 50 mg, 70 mg tvrdé tobolky (enkorafenib) – cytostatikum, inhibitor proteinkináz – rozšíření indikací: nově v kombinaci s binimetinibem k léčbě dospělých pacientů s pokročilým nemalobuněčným karcinomem plic s mutací V600E genu BRAF.
Clonidine Olikla 150 mikrogramů/ml injekční roztok (klonidin-hydrochlorid) – hypertenzní krize a případy hypertenze, kdy je dočasně nemožné perorální podání nebo je toto podání považováno za nedostatečně účinné. Parenterální podání je vyhrazeno pro hospitalizované pacienty.
Comirnaty KP.2 30 mikrogramů/dávku injekční disperze/injekční disperze v předplněné injekční stříkačce, mRNA vakcína proti onemocnění covid-19 (mRNA kódující kódující spike protein viru SARS-CoV-2 - KP.2) – k aktivní imunizaci k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2 u osob ve věku 12 let a starších.
Comirnaty KP.2 10 mikrogramů/dávku injekční disperze, mRNA vakcína proti onemocnění covid-19 (mRNA kódující kódující spike protein viru SARS-CoV-2 - KP.2) – k aktivní imunizaci k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2 u dětí ve věku 5 až 11 let.
Comirnaty KP.2 3 mikrogramy/dávku koncentrát pro injekční disperzi, mRNA vakcína proti onemocnění covid-19 (mRNA kódující kódující spike protein viru SARS-CoV-2 - KP.2) – k aktivní imunizaci k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2 u kojenců a dětí ve věku od 6 měsíců do 4 let.
Vakcínu je třeba používat v souladu s oficiálními doporučeními.
EURneffy 2 mg nosní sprej, roztok v jednodávkovém obalu (epinefrin) – k urgentní léčbě alergických reakcí (anafylaxe) způsobených bodnutím nebo štípnutím hmyzem, potravinami, léčivými přípravky a dalšími alergeny, jakož i idiopatické anafylaxe nebo anafylaxe vyvolané fyzickou námahou. Léčba je určena pro dospělé, dospívající a děti s t.hm. >= 30 kg.
Kayfanda 200, 400, 600 a 12000 mikrogramů tvrdé tobolky (seskvihydrát odevixibátu) – léčivo k terapii onemocnění žlučových cest – léčba cholestatického pruritu u Alagillova syndromu (ALGS) u pacientů od 6 měsíců.
Octagam 50 mg/ml a 100 mg/ml infuzní roztok (normální lidský imunoglobulin pro intravenózní podání) – nová indikace: preexpoziční a postexpoziční profylaxe spalniček u vnímavých dospělých, dětí a dospívajících (0-18 let), u kterých je aktivní imunizace kontraindikována nebo se nedoporučuje.
Ospolot 20 mg/ml perorální suspenze (sultiam) – nová léková forma – rolandická epilepsie (benigní dětská epilepsie s centrotemporálními hroty).
Padcev 20 mg, 30 mg prášek pro koncentrát pro infuzní roztok (enfortumab vedotin) – cytostatikum, konjugát protilátka-léčivo – rozšíření indikace: nově v kombinaci s pembrolizumabem v první linii léčby dospělých pacientů s neresekovatelným nebo metastazujícím uroteliálním karcinomem způsobilých k chemoterapii s platinou.
Panzyga 100 mg/ml infuzní roztok (normální lidský imunoglobulin pro intravenózní podání) – nová indikace: preexpoziční a postexpoziční profylaxe spalniček u vnímavých dospělých, dětí a dospívajících (0-18 let), u kterých je aktivní imunizace kontraindikována nebo se nedoporučuje.
Pelafen perorální roztok (kapky) a sirup (kořen pelargonie) – fytofarmakum – tradiční rostlinný léčivý přípravek k symptomatické léčbě nachlazení u dospělých, dospívajících a dětí od 6 let. Použití je založeno výlučně na zkušenosti z dlouhodobého použití.
Pitilox 1 mg, 2 mg a 4 mg (pitavastatin) – ke snížení zvýšeného celkového cholesterolu (TC) a LDL-C u dospělých, dospívajících a dětí ve věku od 6 let s primární hypercholesterolemií, včetně heterozygotní familiární hypercholesterolemie a kombinované (smíšené) dyslipidemie, pokud odpověď na dietu a jiná nefarmakologická opatření není adekvátní.
Iasophyr 225 ppm, 450 ppm nebo 1000 ppm medicinální plyn, stlačený (oxid dusnatý) – léčba perzistentní plicní hypertenze novorozenců, léčba plicní hypertenze spojené s operací srdce u pacientů všech věkových kategorií.
Ilon mast (blahovičníková silice, modřínový terpentýn, terpentýnová silice) – fytofarmakum – tradiční rostlinný léčivý přípravek používaný k léčbě mírných ohraničených hnisavých zánětů kůže, například zánětlivých uzlíků, folikulitidy a zánětů potních žláz u dospělých, dospívajících a dětí od 6 let. Použití je založeno výlučně na zkušenosti z dlouhodobého použití.
Iqirvo 80 mg potahované tablety (elafibranor) – léčivo k terapii onemocnění žlučových cest – léčba primární biliární cholangitidy (PBC) v kombinaci s kyselinou ursodeoxycholovou (UDCA) u dospělých s nedostatečnou odpovědí na UDCA nebo jako monoterapie u pacientů, kteří netolerují léčbu UDCA.
Loqtorzi 240 mg koncentrát pro infuzní roztok (toripalimab) – cytostatikum, monoklonální protilátka – v kombinaci s cisplatinou a gemcitabinem jako léčba první linie u dospělých pacientů s recidivujícím nebo metastazujícím karcinomem nosohltanu; v kombinaci s cisplatinou a paklitaxelem jako léčba první linie u dospělých pacientů s neresekovatelným pokročilým, recidivujícím nebo metastazujícím spinocelulárním karcinomem jícnu.
Mektovi 15 mg, 45 mg potahované tablety (binimetinib) – cytostatikum, inhibitor proteinkináz – rozšíření indikace: nově v kombinaci s enkorafenibem k léčbě dospělých pacientů s pokročilým nemalobuněčným karcinomem plic s mutací V600E genu BRAF.
Rivaroxaban Zentiva 5 mg/ml perorální suspenze (rivaroxaban) – nová síla perorální suspenze v indikaci: prevence žilního tromboembolismu (VTE) u dospělých pacientů podstupujících elektivní operativní náhradu kyčelního nebo kolenního kloubu; prevence cévní mozkové příhody a systémové embolizace u dospělých pacientů s nevalvulární fibrilací síní s jedním nebo více rizikovými faktory; léčba hluboké žilní trombózy (DVT) a plicní embolie (PE) a prevence recidivující DVT a PE u dospělých; léčba VTE a prevence recidivy VTE u dětí do 18 let s t.hm. od 30 kg po min. 5 dnech úvodní parenterální antikoagulační léčby.
Rybelsus 25 mg a 50 mg tablety (semaglutid) – antidibetikum, GLP-1 agonista – nové síly přípravku k léčbě dospělých s nedostatečně kontrolovaným diabetes mellitus 2. typu ke zlepšení kontroly glykemie jako doplněk k dietním opatřením a cvičení: jako monoterapie, pokud je metformin považován za nevhodný v důsledku nesnášenlivosti nebo kontraindikací; v kombinaci s jinými léčivými přípravky k léčbě diabetu.
Rybrevant 350 mg koncentrát pro infuzní roztok (amivantamab) – cytostatikum, konjugát protilátka-léčivo – rozšíření indikací: nově: v kombinaci s karboplatinou a pemetrexedem k léčbě dospělých pacientů s pokročilým nemalobuněčným karcinomem plic (NSCLC) s delecemi v exonu 19 nebo substitučními mutacemi L858R v exonu 21 genu kódujícího EGFR po selhání předchozí terapie, včetně inhibitoru tyrosinkinázy (TKI) EGFR; v kombinaci s karboplatinou a pemetrexedem k prvoliniové léčbě dospělých pacientů s pokročilým NSCLC s aktivujícími inzerčními mutacemi v exonu 20 genu kódujícího EGFR.
Spikevax JN.1 0,1 mg/ml injekční disperze
Spikevax JN.1 50 mikrogramů injekční disperze
Spikevax JN.1 50 mikrogramů injekční disperze v předplněné injekční stříkačce
mRNA vakcína proti onemocnění covid-19 (mRNA kódující kódující spike protein viru SARS-CoV-2 - JN.1) – k aktivní imunizaci jedinců od 6 měsíců věku k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2. Vakcínu je třeba používat v souladu s oficiálními doporučeními.
Vyloy 100 mg prášek pro koncentrát pro infuzní roztok (zolbetuximab) – cytostatikum, konjugát protilátka-léčivo – v kombinaci s chemoterapií s obsahem fluorpyrimidinu a platiny v první linii léčby dospělých pacientů s lokálně pokročilým neresekovatelným nebo metastazujícím HER2-negativním adenokarcinomem žaludku nebo gastroezofageální junkce (GEJ), který je claudin 18.2 (CLDN18.2) pozitivní.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
16.9.2024
Aktualizace ZÁŘÍ 2024
Aktualizace AISLP září 2024 je připravena ke stažení.
Vážení uživatelé programu AISLP,
zasíláme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální zářijové verzi AISLP.
Abiraterone Sandoz 500 mg, 1000 mg potahované tablety (abirateron-acetát) – cytostatikum – rozšíření indikace: nově také spolu s prednisonem nebo prednisolonem k léčbě nově diagnostikovaného, vysoce rizikového nemetastazujícího hormonálně senzitivního karcinomu prostaty (HSPC) u dospělých mužů v kombinaci s androgen-deprivační léčbou (ADT) a radioterapií.
Adzynma 500 IU a 1500 IU prášek a rozpouštědlo pro injekční roztok (rADAMTS13) – enzymová substituční terapie k léčbě deficitu ADAMTS13 u pacientů s vrozenou trombotickou trombocytopenickou purpurou (cTTP), pro všechny věkové skupiny.
Akantior 0,8 mg/ml oční kapky (polyhexanid) – oftalmologikum – léčba akantamébové keratitidy u dospělých a dospívajících od 12 let.
Alecensa 150 mg tvrdé tobolky (alektinib-hydrochlorid) – cytostatikum, inhibitor proteinkináz – rozšíření indikace: nově také v monoterapii jako adjuvantní léčba po úplné resekci nádoru u dospělých pacientů s ALK pozitivním resekovaný nemalobuněčným karcinomem plic (NSCLC) s vysokým rizikem rekurence.
Atorvastatin/Amlodipine/Ramipril Zentiva 10 mg/5 mg/5 mg tablety; 20 mg/5 mg/5 mg tablety; 20 mg/5 mg/10 mg tablety; 20 mg/10 mg/10 mg tablety a 40 mg/10 mg/10 mg tablety – nová fixní kombinace k léčbě hypertenze spojené s primární hypercholesterolemií nebo smíšenou hyperlipidemií u dospělých jako náhrada léčby u pacientů, kteří jsou již adekvátně kontrolováni atorvastatinem, amlodipinem a ramiprilem podávanými jako samostatné přípravky souběžně ve stejné dávce jako v této kombinaci.
Balversa 3 mg, 4 mg, 5 mg potahované tablety (erdafitinib) – cytostatikum, inhibitor proteinkináz – monoterapie dospělých pacientů s neresekovatelným nebo metastazujícím uroteliálním karcinomem (UC) s citlivými genetickými alteracemi FGFR3, kteří byli léčeni nejméně jednou linií terapie zahrnující inhibitor PD-1 nebo PD-L1 v neresekovatelném nebo metastazujícím stádiu léčby.
Bimzelx 160 mg a 320 mg injekční roztok v předplněné injekční stříkačce/peru (bimekizumab) – nová síla 320 mg: léčba středně těžké až těžké formy ložiskové psoriázy u dospělých, u nichž je indikována systémová léčba. Léčba aktivní psoriatické artritidy u dospělých, kteří reagovali nedostatečně nebo netolerovali jeden nebo více chorobu modifikujících antirevmatických léků (DMARD), v monoterapii nebo v kombinaci s methotrexátem. Léčba dospělých s aktivní neradiografickou axiální spondylartritidou s objektivními známkami zánětu indikovanými zvýšenou hladinou C-reaktivního proteinu a/nebo magnetickou rezonancí, kteří reagovali nedostatečně nebo netolerovali nesteroidní protizánětlivé léky. Léčba dospělých s aktivní ankylozující spondylitidou, kteří reagovali nedostatečně nebo netolerovali konvenční léčbu. Léčba aktivní středně těžké až těžké hidradenitis suppurativa (acne inversa, HS) u dospělých s nedostatečnou reakcí na jinou konvenční systémovou léčbu HS.
Cresemba 200 mg prášek pro koncentrát pro infuzní roztok (isavukonazol) – antimykotikum – rozšíření indikace: přípravek je indikován u pacientů ve věku od 1 roku k léčbě invazivní aspergilózy; mukormykózy u pacientů, kterým nelze podat amfotericin B. Je třeba zohlednit oficiální pokyny ke správnému použití antimykotik.
Cresemba 40 mg a 100 mg tvrdé tobolky (isavukonazol) – antimykotikum – nová síla přípravku a rozšíření indikace: přípravek je indikován u dospělých a pediatrických pacientů ve věku od 6 let k léčbě invazivní aspergilózy; mukormykózy u pacientů, kterým nelze podat amfotericin B. Je třeba zohlednit oficiální pokyny ke správnému použití antimykotik. Tvrdé tobolky 40 mg jsou určeny k použití u pediatrických pacientů.
Eliquis 0,15 mg granule v tobolkách k otevření (apixaban) – nová síla a léková forma k léčbě žilního tromboembolismu (VTE) a prevence rekurence VTE u pediatrických pacientů ve věku od 28 dnů (s t.hm. 4 až <5 kg).
Eliquis 0,5 mg, 1,5 mg a 2 mg obalené granule v sáčku (apixaban) – nová síla a léková forma k léčbě žilního tromboembolismu (VTE) a prevence rekurence VTE u pediatrických pacientů ve věku od 28 dnů (s t.hm. 5 až <35 kg).
Galliapharm 1,11 GBq, 1,48 GBq, 1,85 GBq , 2,22 GBq, 2,59 GBq, 2,96 GBq, 3,33 GBq a 3,70 GBq radionuklidový generátor (chlorid germaničitý-(68Ge), chlorid gallitý-(68Ga)) – diagnostické radiofarmakum – in vitro značení kitů pro radiofarmaka, které byly vyvinuty a schváleny k radioaktivnímu značení tímto roztokem, a to k zobrazování pomocí pozitronové emisní tomografie (PET).
mRESVIA injekční disperze v předplněné injekční stříkačce, mRNA vakcína (modifikovaný nukleosid) proti respiračnímu syncytiálnímu viru (RSV), zapouzdřené v lipidových nanočásticích, léčivou látkou je jednovláknová mRNA s čepičkou na 5' konci kódující glykoprotein F viru RSV-A stabilizovaný v prefuzní konformaci – k aktivní imunizaci k prevenci onemocnění dolních cest dýchacích způsobeného respiračním syncytiálním virem (RSV) u dospělých ve věku 60 let a starších. Vakcína se má používat v souladu s oficiálními doporučeními.
Ordspono 2 mg, 80 mg, 320 mg koncentrát pro infuzní roztok (odronextamab) – cytostatikum, monoklonální protilátka – monoterapie dospělých pacientů s relabujícím nebo refrakterním folikulárním lymfomem (r/r FL) po dvou nebo více liniích systémové léčby.
Piasky 340 mg injekční/infuzní roztok (krovalimab) – monoterapie paroxysmální noční hemoglobinurie (PNH) u dospělých a pediatrických pacientů ve věku 12 let a starších s t.hm. 40 kg a vyšší: s hemolýzou s klinickými příznaky svědčícími o vysoké aktivitě onemocnění nebo u pacientů, kteří jsou klinicky stabilní po léčbě inhibitorem složky 5 komplementu (C5) po dobu nejméně posledních 6 měsíců.
Plexyl roztok pro kardioplegii (síran hořečnatý, chlorid draselný, xylitol, prokain-hydrochlorid) – k vyvolání okamžité a prodloužené diastolické kardioplegické zástavy u dospělých pacientů.
Pulfunvu kit pro radiofarmakum (grafit) – diagnostické radiofarmakum – scintigrafie alveolárních prostorů, zejména v kontextu s diagnostikou plicní embolie.
Rybelsus 1,5 mg, 4 mg a 9 mg tablety (semaglutid) – antidibetikum, GLP-1 agonista – nové síly přípravku k léčbě dospělých s nedostatečně kontrolovaným diabetes mellitus 2. typu ke zlepšení kontroly glykemie jako doplněk k dietním opatřením a cvičení: jako monoterapie, pokud je metformin považován za nevhodný v důsledku nesnášenlivosti nebo kontraindikací; v kombinaci s jinými léčivými přípravky k léčbě diabetu.
Steqeyma 45 mg a 90 mg injekční roztok v předplněné injekční stříkačce, 130 mg koncentrát pro infuzní roztok (ustekinumab) – biosimilar.
Tauvid 800 MBq/ml a 1900 MBq/ml injekční roztok (flortaucipir-(18F)) – diagnostické radiofarmakum – vyšetření mozku pozitronovou emisní tomografií (PET) za účelem posouzení přítomnosti neokortikálních agregátů neurofibrilárních klubek (NFT) tau proteinu u dospělých pacientů s kognitivním deficitem, kteří jsou vyšetřováni pro Alzheimerovu nemoc (AD). Vyšetření pomocí flortaucipiru-(18F) je doplňkem klinického vyšetření a dalších diagnostických metod.
Winrevair 45 mg a 60 mg prášek pro injekční roztok
Winrevair 45 mg a 60 mg prášek a rozpouštědlo pro injekční roztok (sotatercept) – k léčbě plicní arteriální hypertenze (PAH) v kombinaci s jinými terapiemi PAH u dospělých pacientů s funkční třídou dle WHO (WHO FC) II až III ke zlepšení zátěžové kapacity.
Xalkori 200 mg a 250 mg tvrdé tobolky
Xalkori 20 mg, 50 mg a 150 mg granule v tobolkách k otevření (krizotinib) – cytostatikum, inhibitor tyrozinkináz – nová léková forma přípravku a rozšíření indikace: nově také k léčbě pediatrických pacientů od 1 roku relabujícím nebo refrakterním systémovým anaplastickým velkobuněčným lymfomem (ALCL) pozitivním nakinázu anaplastického lymfomu (ALK); s rekurentním nebo refrakterním, neresekovatelným zánětlivým myofibroblastickým nádorem (IMT) pozitivním na kinázu anaplastického lymfomu (ALK).
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
13.9.2024
SÚKL informuje o možnostech, jak zajistit léky v případech, kdy není možné navštívit lékaře osobně, například během povodní nebo jiných krizových situací.
10.9.2024
Informace o přípravku budou aktualizovány za účelem zvýšení povědomí o známém riziku agranulocytózy a usnadnění jeho včasného rozpoznání a diagnostiky.
6.9.2024
SÚKL informuje o stažení uvedené šarže léčivého přípravku Temozolomide Glenmark, 100 mg cps.dur. 5 až z úrovně pacientů.
6.9.2024
Státní ústav pro kontrolu léčiv informuje o závadě v jakosti léčivého přípravku Temozolomide Glenmark, který je používán k léčbě specifických forem mozkových nádorů.
6.9.2024
Informace o závadách v jakosti, opatření z registračních důvodů a nežádoucích účincích léčiv, padělcích a nelegálních přípravcích.
30.8.2024
SÚKL informuje o stažení uvedené šarže léčivého přípravku Imunor, 10mg por. lyo. 4 až z úrovně zdravotnických zařízení.
21.8.2024
SÚKL informuje o uvolnění distribuce, výdeje a léčebného použití uvedené šarže léčivého přípravku NovoSeven, 2mg(100KIU) inj. pso. lqf. 1+1X2ml III.
13.8.2024
Aktualizace SRPEN 2024
Aktualizace AISLP srpen 2024 je připravena ke stažení.
Vážení uživatelé programu AISLP,
zasíláme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální srpnové verzi AISLP.
Agilus 120 mg prášek pro injekční roztok (dantrolen) – léčba maligní hypertermie u dospělých a dětí všech věkových kategorií v kombinaci s odpovídajícími podpůrnými opatřeními.
Alutard SQ bojínek injekční suspenze
Alutard SQ bříza injekční suspenze
Alutard SQ kočka injekční suspenze
Alutard SQ pes injekční suspenze
Alutard SQ roztoči injekční suspenze
depotní přípravek obsahující standardizovaný extrakt alergenů – změna názvu přípravku (dříve Alutard SQ), aktualizace textů SmPC a PIL.
Canagliflozin Teva 100 mg a 300 mg potahované tablety – antidiabetikum, inhibitor SGLT2 – první generikum s léčivou látkou kanagliflozin: k léčbě dospělých s nedostatečně kontrolovaným diabetes mellitus 2. typu jako přídatná terapie k dietě a cvičení: v monoterapii, pokud se metformin považuje za nevhodný v důsledku nesnášenlivosti nebo kontraindikací; jako přídatná léčba k dalším léčivým přípravkům k léčbě diabetu.
Cejemly 600 mg koncentrát pro infuzní roztok (sugemalimab) – cytostatikum, monoklonální protilátka – v kombinaci s chemoterapií na bázi platiny v první linii léčby dospělých s metastazujícím nemalobuněčným karcinomem plic (NSCLC) bez senzibilizujících mutací EGFR a bez aberací genomu ALK, ROS1 a RET.
Comirnaty JN.1 30 mikrogramů/dávku injekční disperze/injekční disperze v předplněné injekční stříkačce, mRNA vakcína proti onemocnění covid-19 (bretovameran) – k aktivní imunizaci k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2 u osob ve věku 12 let a starších.
Comirnaty JN.1 10 mikrogramů/dávku injekční disperze, mRNA vakcína proti onemocnění covid-19 (bretovameran) – k aktivní imunizaci k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2 u dětí ve věku 5 až 11 let.
Comirnaty JN.1 10 mikrogramů/dávku koncentrát pro injekční disperzi, mRNA vakcína proti onemocnění covid-19 (bretovameran) – k aktivní imunizaci k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2 u dětí ve věku 5 až 11 let.
Comirnaty JN.1 3 mikrogramy/dávku koncentrát pro injekční disperzi, mRNA vakcína proti onemocnění covid-19 (bretovameran) – k aktivní imunizaci k prevenci onemocnění covid-19 způsobeného virem SARS-CoV-2 u kojenců a dětí ve věku od 6 měsíců do 4 let.
Vakcínu je třeba používat v souladu s oficiálními doporučeními.
Crysvita 10 mg, 20 mg a 30 mg injekční roztok v předplněné injekční stříkačce (burosumab) – nová léková forma – léčba X-vázané hypofosfatemie (X-Linked Hypophosphataemia, XLH) u dětí a dospívajících ve věku od 1 do 17 let s rentgenograficky prokázanou nemocí kostí a u dospělých. Léčba hypofosfatemie spojené s FGF23 (růstovým faktorem fibroblastů 23) u dětí a dospívajících ve věku od 1 roku do 17 let a dospělých s osteomalacií indukovanou tumorem (tumour-induced osteomalacia, TIO), spojenou s fosfaturickými mesenchymálními tumory, u kterých nelze provést kurativní resekci či lokalizaci.
Dupixent 300 mg 300 mg injekční roztok v předplněné injekční stříkačce/předplněném peru (dupilumab) – rozšíření indikací o CHOPN: přípravek je indikován u dospělých pacientů jako přídatná udržovací léčba nekontrolované chronické obstrukční plicní nemoci (CHOPN) charakterizované zvýšeným počtem eozinofilů v krvi, v kombinaci s inhalačním kortikosteroidem (IKS), dlouhodobě působícím beta2-agonistou (LABA) a dlouhodobě působícím muskarinovým antagonistou (LAMA) nebo v kombinaci s LABA a LAMA, nejsou-li IKS vhodné.
Durveqtix 0,79-1,21 x 10 na 13 vektorových genomů/ml koncentrát pro infuzní roztok (fidanakogen elaparvovek) – genová terapie dospělých pacientů s těžkou a středně těžkou hemofilií B bez inhibitorů faktoru IX v anamnéze a bez detekovatelných protilátek proti variantě AAV sérotypu Rh74.
Efluelda Tetra injekční suspenze v předplněné injekční stříkačce, tetravalentní vakcína proti chřipce (štěpený virion, inaktivovaný), 60 mikrogramů HA/kmen
Fluad Tetra injekční suspenze v předplněné injekční stříkačce, vakcína proti chřipce (povrchový antigen, inaktivovaná, adjuvovaná)
Fluarix Tetra injekční suspenze v předplněné injekční stříkačce, vakcína proti chřipce (štěpený virion, inaktivovaný)
Flucelvax Tetra injekční suspenze v předplněné injekční stříkačce, vakcína proti chřipce (povrchový antigen, inaktivovaná, připravená na buněčných kulturách)
Fluenz nosní sprej, suspenze, vakcína proti chřipce (živá atenuovaná, nosní)
Influvac Tetra injekční suspenze v předplněné injekční stříkačce, vakcína proti chřipce (povrchový antigen, inaktivovaná) aktualizace složení vakcín proti sezónní chřipce dle doporučení Světové zdravotnické organizace (WHO) pro severní polokouli a doporučení Evropské unie pro sezónu 2024/2025.
Ixchiq prášek a rozpouštědlo pro injekční roztok, vakcína proti onemocnění Chikungunya (živý oslabený Chikungunya virus (CHIKV), kmen delta5nsP3, produkovaný ve Vero buňkách) – k aktivní imunizaci k prevenci onemocnění způsobeného Chikungunya virem (CHIKV) u osob ve věku 18 let a starších. Vakcínu je třeba používat v souladu s oficiálními doporučeními.
Jeraygo 12,5 mg a 25 mg potahované tablety (aprocitentan) – antihypertenzivum – k léčbě rezistentní hypertenze u dospělých pacientů v kombinaci s nejméně třemi dalšími antihypertenzivy.
Livmarli 9,5 mg/ml perorální roztok (maralixibát) – léčivo k terapii onemocnění žlučových cest – rozšíření indikace: léčba progresivní familiární intrahepatální cholestázy (PFIC) u pacientů od 3 měsíců.
Obgemsa 75 mg potahované tablety (vibegron) – urologikum – symptomatická léčba dospělých pacientů se syndromem hyperaktivního močového měchýře (OAB).
Rozlytrek 100 mg a 200 mg tvrdé tobolky, 50 mg potahované granule v sáčku (entrektinib) – cytostatikum, inhibitor proteinkináz – rozšíření indikace: v monoterapii k léčbě dospělých a pediatrických pacientů od 1 měsíce se solidními nádory s fúzí genu NTRK.
Skyrizi 600 mg koncentrát pro infuzní roztok a 90 mg injekční roztok v předplněné injekční stříkačce (risankizumab) – rozšíření indikací: Crohnova choroba: léčba dospělých pacientů se středně těžkou až těžkou aktivní Crohnovou chorobou s nedostatečnou odpovědí, se ztrátou odpovědi nebo intolerancí konvenční léčby nebo biologické léčby. Ulcerózní kolitida: léčba dospělých pacientů se středně těžkou až těžkou aktivní ulcerózní kolitidou s nedostatečnou odpovědí, se ztrátou odpovědi nebo intolerancí konvenční léčby nebo biologické léčby.
Skyrizi 180 mg a 360 mg injekční roztok v zásobní vložce (risankizumab) – nová síla přípravku (180 mg) a rozšíření indikací: Crohnova choroba: léčba dospělých pacientů se středně těžkou až těžkou aktivní Crohnovou chorobou s nedostatečnou odpovědí, se ztrátou odpovědi nebo intolerancí konvenční léčby nebo biologické léčby. Ulcerózní kolitida: léčba dospělých pacientů se středně těžkou až těžkou aktivní ulcerózní kolitidou s nedostatečnou odpovědí, se ztrátou odpovědi nebo intolerancí konvenční léčby nebo biologické léčby.
Vabinxo 160 mg/1,5 mg tablety s řízeným uvolňováním
Valsartan/Indapamid Krka 160 mg/1,5 mg tablety s řízeným uvolňováním
Vamipino 160 mg/1,5 mg tablety s řízeným uvolňováním (valsartan, indapamid) – nová fixní kombinace k léčbě esenciální hypertenze jako náhrada terapie u dospělých pacientů adekvátně kontrolovaných valsartanem a indapamidem podávanými současně ve stejné dávce jako v kombinaci, ale jako samostatné tablety.
Zegalogue 0,6 mg injekční roztok v předplněném peru/injekční stříkačce (dasiglukagon) – hormon – léčba těžké hypoglykemie u dospělých, dospívajících a dětí od 6 let s diabetes mellitus.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
1.8.2024
Letní prodejní akce
I v srpnu ještě můžete využít naší probíhající letní akci!

Akce platí v období od 1.7.2024 do 31.8.2024 včetně a platí pro nové zákazníky. Společnost INPHARMEX, spol. s r.o. si vyhrazuje právo změnit délku trvání nebo podmínky akce.
Více informací na www.aislp.cz, kliknutím na reklamní proužek.
Váš tým AISLP
www.aislp.cz
31.7.2024
SÚKL informuje o stažení uvedené šarže léčivého přípravku Imunor, 10mg por. lyo. 4 až z úrovně zdravotnických zařízení.
29.7.2024
Státní ústav pro kontrolu léčiv informuje,že použití opioidních léků s léčivým přípravkem Mysimba může vést k vážným nežádoucím účinkům.
26.7.2024
SÚKL informuje o pozastavení distribuce a výdeje uvedené šarže léčivého přípravku Temozolomide Glenmark, 100 mg cps. dur. 5.
22.7.2024
SÚKL informuje o stažení uvedených šarží léčivých přípravků Atomoxetin Glenmark, 10mg cps. dur. 28 I, Atomoxetin Glenmark, 18mg cps. dur. 28 I, Atomoxetin Glenmark, 25mg cps. dur. 28 I, Atomoxetin Glenmark, 40mg cps. dur. 28 I a Atomoxetin Glenmark, 60mg cps. dur. 28 I až z úrovně zdravotnických zařízení.
19.7.2024
SÚKL informuje o stažení uvedených šarží léčivých přípravků Atofab, 10 mg cps. dur. 30 i, Atofab, 18 mg cps. dur. 30 I, Atofab, 25 mg cps. dur. 30 I a Atofab, 40 mg cps. dur. 30 I až z úrovně zdravotnických zařízení.
17.7.2024
Od zavedení povinné elektronické preskripce v roce 2018 lékaři vystavili již více než pět set milionů eReceptů. Elektronický předpis s pořadovým číslem 500 000 000 byl vystaven 17. července 2024 po 9. hodině ve Slavkově u Brna.
16.7.2024
SÚKL informuje o stažení uvedených šarží léčivých přípravků Atomoxetin Sandoz 100mg cps. dur. 28 I, Atomoxetin Sandoz 40mg cps. dur. 28 I, Atomoxetin Sandoz 18mg cps. dur. 28 I a Atomoxetin Sandoz 80mg cps. dur. 28 I až z úrovně zdravotnických zařízení.
15.7.2024
SÚKL informuje o stažení uvedené šarže léčivého přípravku Bitinex, 25mg cps. dur. 28 I, až z úrovně zdravotnických zařízení.
13.7.2024
Aktualizace ČERVENEC 2024
Aktualizace AISLP červenec 2024 je připravena ke stažení.
Vážení uživatelé programu AISLP,
zasíláme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální červencové verzi AISLP.
Altuvoct 250 IU, 500 IU, 750 IU, 1000 IU, 2000 IU, 3000 IU, 4000 IU prášek a rozpouštědlo pro injekční roztok (efanesoktokog alfa) – rekombinantní fúzní protein (rFVIII-Fc-vWF-XTEN) k léčbě a prevenci krvácení u pacientů s hemofilií A, pro všechny věkové skupiny.
Benzylpenicillin sodium Kabi 1 MIU, 5 MIU a 10 MIU prášek pro injekční/infuzní roztok (sodná sůl benzylpenicilinu) – přípravek je indikován u dospělých, dospívajících, dětí, novorozenců a předčasně narozených, k léčbě následujících infekcí způsobených citlivými mikroorganismy: akutní bakteriální infekce kůže a kožních struktur (ABSSI), difterie (kromě antitoxinu), komunitní pneumonie, empyém, bakteriální endokarditida, peritonitida, meningitida, abscesy v mozku, osteomyelitida, infekce genitálního ústrojí způsobené fusobakteriemi, gonorea (gonoroická endokarditida nebo artritida), syfilis (kongenitální syfilis), Lymeská borrelióza (meningopolyneuritida Garin-Bujadoux-Bannwarth, acrodermatitis chronica atrophicans, lymeská artritida, lymeská karditida). Přípravek může být také používán k léčbě následujících specifických infekcí: antrax, tetanus, plynatá sněť, listerióza, pasteurelóza, horečka z krysího kousnutí, fusospirochetóza, aktinomykóza. Je třeba vzít v úvahu oficiální pokyny pro vhodné použití antibakteriálních látek.
Carvykti infuzní disperze CAR-pozitivních životaschopných T-lymfocytů (ciltakabtegen autoleucel) – cytostatikum – změna indikace: léčba dospělých pacientů s relabujícím a refrakterním mnohočetným myelomem, kteří podstoupili alespoň jednu předchozí terapii zahrnující imunomodulátor a inhibitor proteazomu, a kteří během poslední léčby vykázali progresi onemocnění a jsou refrakterní na lenalidomid.
Ephedrine Kabi 10 mg/ml injekční roztok (efedrin-hydrochlorid) – sympatomimetikum – léčba hypotenze při spinální, epidurální a celkové anestezii u dospělých a dětí (starších 12 let).
Fluenz nosní sprej, suspenze; vakcína proti chřipce (živá atenuovaná, nosní) – trivalentní vakcína proti chřipce k intranazální aplikaci: profylaxe chřipky u dětí a dospívajících od 24 měsíců do méně než 18 let věku. Přípravek se má používat na základě oficiálních doporučení.
Fruzaqla 1 mg, 5 mg (fruchintinib) – cytostatikum, inhibitor tyrozinkináz – monoterapie dospělých pacientů s metastazujícím kolorektálním karcinomem (mCRC), kteří byli dříve léčeni dostupnými typy léčby, včetně chemoterapie na bázi fluorpyrimidinu, oxaliplatiny a irinotekanu, anti-VEGF látek a anti-EGFR látek, a u kterých došlo k progresi nebo netolerují léčbu trifluridinem s tipiracilem nebo regorafenibem.
Ocrevus 920 mg injekční roztok (okrelizumab) – imunosupresivum, monoklonální protilátka – nová léková forma a síla přípravku – léčba dospělých pacientů s relabujícími formami roztroušené sklerózy (RRS) s aktivním onemocněním definovaným klinicky nebo pomocí zobrazovacích metod. Léčba dospělých pacientů s časnou primárně progresivní roztroušenou sklerózou (PPRS) s ohledem na délku trvání onemocnění, stupeň disability a zánětlivou aktivitou prokázanou zobrazovacími metodami.
Onivyde pegylated liposomal 4,3 mg/ml koncentrát pro infuzní disperzi (irinotekan-sukrosofát) – cytostatikum – rozšíření indikace: léčba metastazujícího adenokarcinomu pankreatu u dospělých pacientů v kombinaci s oxaliplatinou, fluoruracilem (5-FU) a leukovorinem (LV) v první linii.
Qalsody 100 mg injekční roztok (tofersen) – antisense oligonukleotid – léčba dospělých pacientů s amyotrofickou laterální sklerózou (ALS), která je spojena s mutací genu pro superoxid dismutázu 1 (SOD1).
Truqap 160 mg, 200 mg potahované tablety (kapivasertib) – cytostatikum – v kombinaci s fulvestrantem k léčbě dospělých pacientů s lokálně pokročilým nebo metastazujícím karcinomem prsu s pozitivitou estrogenových receptorů (ER), negativitou receptorů pro lidský epidermální růstový faktor typu 2 (HER-2) a s jednou nebo více alteracemi PIK3CA/AKT1/PTEN, po rekurenci nebo progresi při/po hormonální terapii.
Wezenla 130 mg koncentrát pro infuzní roztok (ustekinumab) – biosimilar, imunosupresivum, inhibitor interleukinu – léčba dospělých pacientů se středně těžkou až těžkou aktivní Crohnovou chorobou, u kterých buď odpověď na konvenční terapii nebo na antagonistu tumor nekrotizujícího faktoru alfa (TNF-alfa) nebyla dostatečná nebo odezněla nebo tito pacienti netolerovali konvenční léčbu nebo léčbu TNF-alfa, případně jsou u nich tyto terapie kontraindikovány.
Wezenla 45 mg a 90 mg injekční roztok/injekční roztok v předplněné injekční stříkačce (ustekinumab) – biosimilar, imunosupresivum, inhibitor interleukinu – léčba středně těžké až těžké plakové psoriázy u dospělých, u kterých selhaly jiné systémové léčby, včetně podávání cyklosporinu, methotrexátu (MTX) a PUVA (psoralen a ultrafialové záření A), nebo kteří tyto léčby nesnášejí nebo jsou u nich kontraindikovány. Léčba středně těžké až těžké plakové psoriázy u dětí a dospívajících pacientů ve věku od 6 let a starších, kteří nejsou pod dostatečnou kontrolou nebo nesnášejí jiné systémové léčby nebo fototerapii. Samostatně nebo v kombinaci s MTX k léčbě aktivní psoriatické artritidy u dospělých pacientů, pokud odpověď na předchozí nebiologické onemocnění modifikující antirevmatické přípravky (DMARD) nebyla dostatečná. Léčba dospělých pacientů se středně těžkou až těžkou aktivní Crohnovou chorobou, u kterých buď odpověď na konvenční terapii nebo na antagonistu tumor nekrotizujícího faktoru alfa (TNF-alfa) nebyla dostatečná nebo odezněla nebo tito pacienti netolerovali konvenční léčbu nebo léčbu TNF-alfa, případně jsou u nich tyto terapie kontraindikovány.
Xtandi 40 mg měkké tobolky, 40 mg a 80 mg potahované tablety (enzalutamid) – cytostatikum, inhibitor signalizace androgenního receptoru – rozšíření indikace: v monoterapii nebo v kombinaci s androgen deprivační léčbou k léčbě dospělých mužů s vysoce rizikovým biochemicky rekurentním (BCR) nemetastazujícím hormon senzitivním karcinomem prostaty (nmHSPC), kteří nejsou vhodní k záchranné radioterapii.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
12.7.2024
SÚKL informuje o stažení uvedených šarží léčivých přípravků Atomoxetin Glenmark, 25mg cps. dur. 28 I, Atomoxetin Glenmark, 18mg cps. dur. 28 I, Atomoxetin Glenmark, 10mg cps. dur. 28 I a Atomoxetin Glenmark, 40mg cps. dur. 28 I až z úrovně zdravotnických zařízení.
9.7.2024
SÚKL informuje o stažení uvedených šarží léčivých přípravků Bitinex, 18mg cps. dur. 28 I, Bitinex, 18mg cps. dur. 56 I a Bitinex, 25mg cps. dur. 28 I až z úrovně zdravotnických zařízení.
4.7.2024
Informace o závadách v jakosti, opatřeních z registračních důvodů a nežádoucích účincích léčiv, padělcích a nelegálních přípravcích.
3.7.2024
Státní ústav pro kontrolu léčiv informuje pacienty a zdravotnické pracovníky o dlouhodobé nedostupnosti léčivého přípravku ENDIARON 250MG TBL FLM 20 (kód SÚKL: 258019), na jehož nedostupnost jsou před prázdninami (s ohledem na jeho oblibu pro léčbu cestovatelských průjmů) stálé dotazy.
2.7.2024
Letní prodejní akce
Užijte si prázdniny s AISLP a vyražte na cesty se svou oblíbenou licencí.

Akce platí v období od 1.7.2024 do 31.8.2024 včetně a platí pro nové zákazníky. Společnost INPHARMEX, spol. s r.o. si vyhrazuje právo změnit délku trvání nebo podmínky akce.
Více informací na www.aislp.cz, kliknutím na reklamní proužek.
Váš tým AISLP
www.aislp.cz
1.7.2024
SÚKL informuje o stažení uvedené šarže léčivého přípravku Bitinex, 25mg cps. dur. 28 I, až z úrovně zdravotnických zařízení.
28.6.2024
SÚKL informuje o stažení uvedené šarže léčivého přípravku Imunor, 10mg por. lyo. 4 až z úrovně zdravotnických zařízení.
28.6.2024
SÚKL informuje o stažení uvedených šarží léčivých přípravků Atomoxetin Glenmark, 25mg cps. dur. 28 I a Atomoxetin Glenmark, 18mg cps. dur. 28 I až z úrovně zdravotnických zařízení.
28.6.2024
SÚKL informuje o stažení uvedených šarží léčivého přípravku Tralgit SR, 200 mg tbl. pro. 30 až z úrovně zdravotnických zařízení.
27.6.2024
SÚKL informuje o stažení uvedené šarže léčivého přípravku Cinacalcet Reddy, 30 mg tbl. flm. 28 až z úrovně zdravotnických zařízení.
26.6.2024
Ministerstvo zdravotnictví ČR (MZ), ve spolupráci se Státním ústavem pro kontrolu léčiv (SÚKL), na základě dosavadních zkušeností s dynamikou lékového trhu, připravilo novelu zákona o léčivech, která vstoupila v platnost dne 1. června 2024. Tento zákon zavádí nové povinnosti pro držitele rozhodnutí o registraci léčiv, distributory, lékárny a pro orgány státní správy. Hlavním cílem je zajistit dostupnost léčivých přípravků pro pacienty.
26.6.2024
SÚKL informuje o stažení uvedené šarže léčivého přípravku Procto-glyvenol, 400mg/40mg sup 10 až z úrovně zdravotnických zařízení.
26.6.2024
SÚKL informuje o stažení uvedené šarže léčivého přípravku NovoSeven, 2mg(100KIU) inj. pso. lqf. 1+1X2ml III až z úrovně distributorů.
26.6.2024
25.6.2024
SÚKL informuje o stažení uvedené šarží léčivého přípravku Atomoxetin Accord, 40mg cps. dur. 28 I až z úrovně zdravotnických zařízení.
21.6.2024
Státní ústav pro kontrolu léčiv informuje o závadě v jakosti léčivého přípravku ACIDI BORCI AQUA OPHTHALMICA C. th. Ten se používá při očních potížích, jako jsou např. lehčí formy zánětů spojivek, dále při pálení, řezání a svědění oka způsobené zevními vlivy. Léčivý přípravek je stahován z důvodu preventivního opatření.
21.6.2024
SÚKL informuje o stažení uvedené šarže léčivého přípravku Acidi borici aqua ophthalmica C. th., 20 g až z úrovně pacientů.
20.6.2024
Státní ústav pro kontrolu léčiv si Vás dovoluje informovat o zahájení evropského přehodnocení. Přehodnocení je zaměřeno na riziko agranulocytózy a na opatření, jak je minimalizovat.
18.6.2024
SÚKL informuje o stažení uvedených šarží léčivých přípravků Atomoxetin Accord, 40mg cps. dur. 28 I a Atomoxetin Accord, 10mg cps. dur. 28 I, až z úrovně zdravotnických zařízení.
12.6.2024
Aktualizace ČERVEN 2024
Aktualizace AISLP červen 2024 je připravena ke stažení.
Vážení uživatelé programu AISLP,
zasíláme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální červnové verzi AISLP.
Amgevita 20 mg, 40 mg a 80 mg injekční roztok v předplněné injekční stříkačce/peru (adalimumab) – imunosupresivum, inhibitor tumor nekrotizujícího faktoru alfa – nové síly přípravku.
Awiqli 700 jednotek/ml injekční roztok v předplněném peru (insulin-ikodek) – antidiabetikum, bazální inzulin k subkutánnímu podání jednou týdně – léčba diabetes mellitus u dospělých.
BCG medac prášek a rozpouštědlo pro intravezikální suspenzi (Bacillus Calmete-Guerin) – imunostimulans – léčba neinvazivního karcinomu urotelu močového měchýře: kurativní léčba karcinomu in situ; profylaktická léčba rekurence urotelového karcinomu omezeného na mukózu (Ta G1-G2 při multifokálním a/nebo rekurentním nádoru a Ta G3), urotelového karcinomu v lamina propria nezasahujícího do svalové tkáně močového měchýře (T1) a karcinomu in situ.
Fabhalta 200 mg tvrdé tobolky (iptakopan) – imunosupresivum, inhibitor komplementu – monoterapie dospělých pacientů s paroxysmální noční hemoglobinurií (PNH), kteří mají hemolytickou anemii.
Jubbonti 60 mg injekční roztok v předplněné injekční stříkačce (denosubam) – biosimilar – léčba osteoporózy u postmenopauzálních žen a u mužů se zvýšeným rizikem zlomenin. U postmenopauzálních žen přípravek významně snižuje riziko zlomenin obratlů, nevertebrálních zlomenin a zlomenin proximálního femuru. Léčba úbytku kostní hmoty vzniklé následkem hormonální ablace u mužů s rakovinou prostaty, u kterých je riziko vzniku zlomenin zvýšeno. U mužů s rakovinou prostaty, léčených hormonální ablací, přípravek významně snižuje riziko zlomenin obratlů. Léčba úbytku kostní hmoty spojeného s dlouhodobou systémovou léčbou glukokortikoidy u dospělých pacientů se zvýšeným rizikem zlomenin.
Lytenava 125 mg/ml injekční roztok (bevacizumab gama) – oftalmologikum – léčba dospělých pacientů s neovaskulární (vlhkou) věkem podmíněnou makulární degenerací (nAMD).
Neoatricon 1,5 mg/ml infuzní roztok
Neoatricon 4,5 mg/ml infuzní roztok (dopamin-hydrochlorid) – léčba hypotenze u hemodynamicky nestabilních novorozenců, kojenců a dětí mladších 18 let.
Retsevmo 40 mg, 80 mg tvrdé tobolky (selperkatinib) – cytostatikum, inhibitor tyrozinkináz – rozšíření indikace: monoterapie dospělých s pokročilými solidními nádory s pozitivitou RET fúze, kdy možnosti léčby necílící na RET poskytují omezený klinický přínos nebo byly vyčerpány.
Vaxigrip Tetra injekční suspenze v předplněné injekční stříkačce, tetravalentní vakcína proti chřipce (štěpený virion, inaktivovaný) – aktualizace složení vakcíny proti sezónní chřipce dle doporučení Světové zdravotnické organizace (WHO) pro severní polokouli a doporučení Evropské unie pro sezónu 2024/2025.
Wyost 120 mg injekční roztok (denosumab) – biosimilar – prevence kostních příhod (skeletal related events, SRE) (patologické fraktury, míšní komprese, stavy vyžadující radiační léčbu kostí či kostní operaci) u dospělých s pokročilými malignitami postihujícími kosti. Léčba velkobuněčného kostního nádoru, který je neresekabilní nebo kde chirurgická resekce povede pravděpodobně k závažné morbiditě, u dospělých a dospívajících s vyvinutým skeletem.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
12.6.2024
SÚKL informuje o pozastavení distribuce, výdeje a léčebného používání uvedené šarže léčivého přípravku NovoSeven, 2mg(100KIU) inj. pso. lqf. 1+1X2ml III.
4.6.2024
Zobrazení grafů vývoje cen volně prodejných léčivých přípravků
Zobrazení grafů vývoje cen volně prodejných léčivých přípravků
Vážení uživatelé AISLP,
rádi bychom Vám představili další novinku, která Vám zpříjemní práci s programem AISLP. V minulých příspěvcích jsme Vás seznámili se zobrazením grafů výdeje léčivých přípravků. Nyní si nově můžete zobrazit i graf vývoje ceny volně prodejných léčivých přípravků v internetových obchodech.
Tato funkce je dostupná jak uživatelům licence Windows, tak těm, kteří využívají internetovou nebo intranetovou verzi AISLP.
Jaká data lze v grafu zobrazit naleznete na následujících obrázcích.
Zobrazení průměrné ceny
Kliknutím na ikonku průměru se zobrazí linka průměrné ceny volně prodejného léčivého přípravku za Vámi zvolené období.
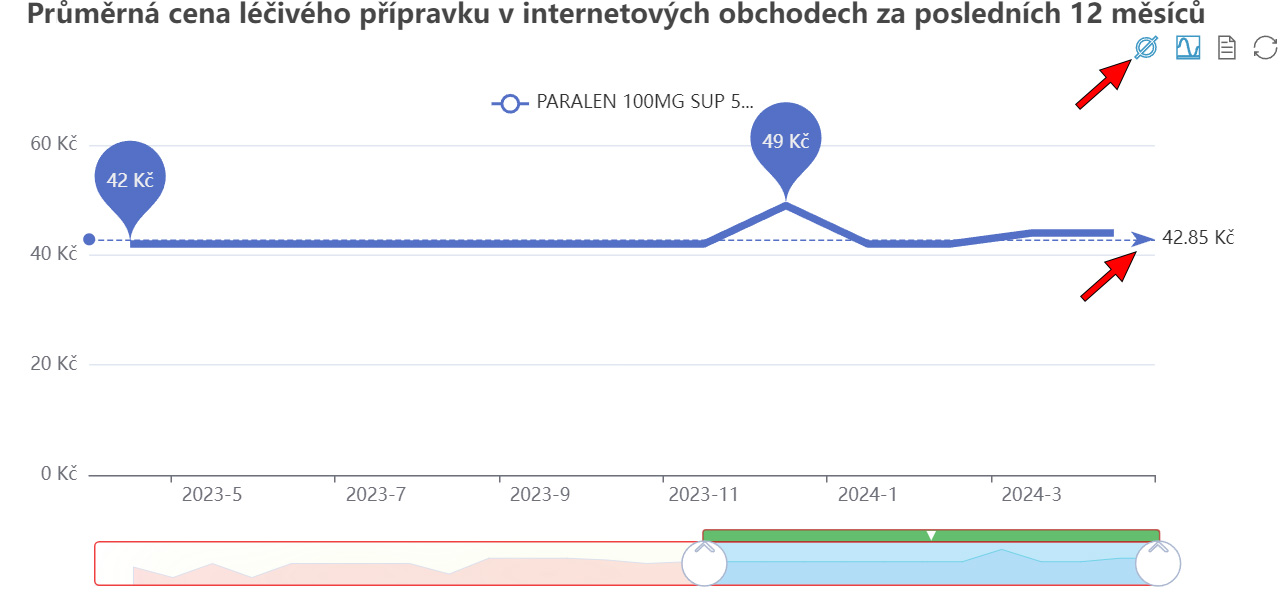
Zobrazení nejvyšší a nejnižší ceny
Kliknutím na ikonku grafu zobrazíte nejnižší a nejvyšší cenu volně prodejného léčivého přípravku za Vámi zvolené období.
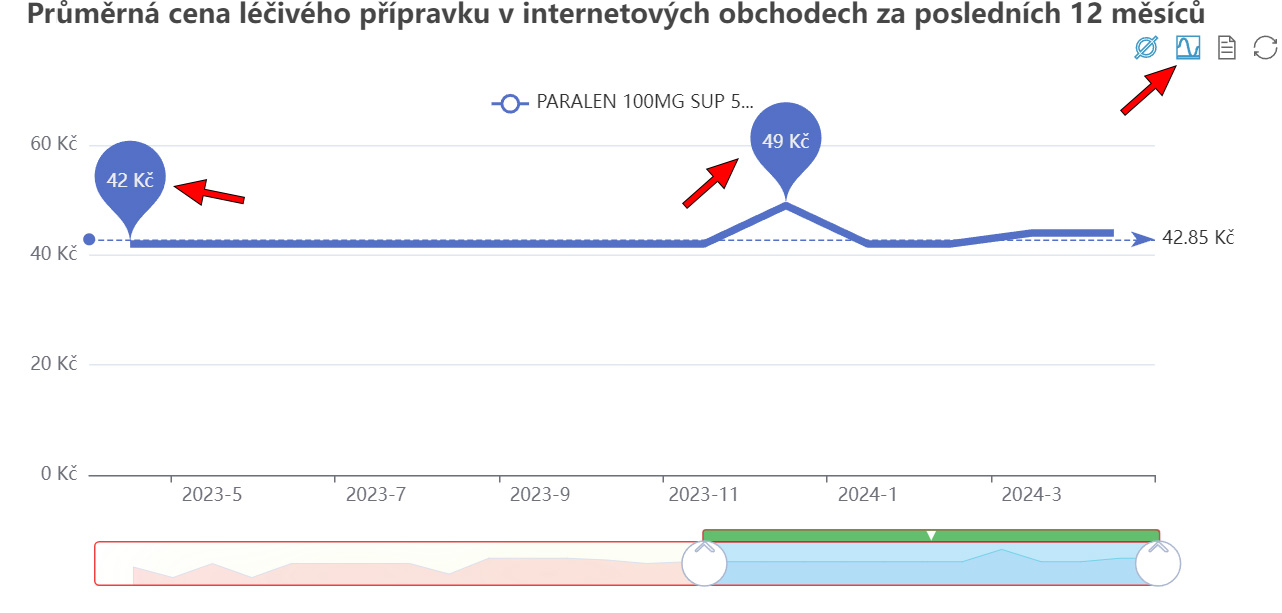
Zobrazení konkrétního údaje
Pokud najedete kurzorem na čáru grafu, zobrazí se vám přesný údaj s cenou pro konkrétní měsíc.
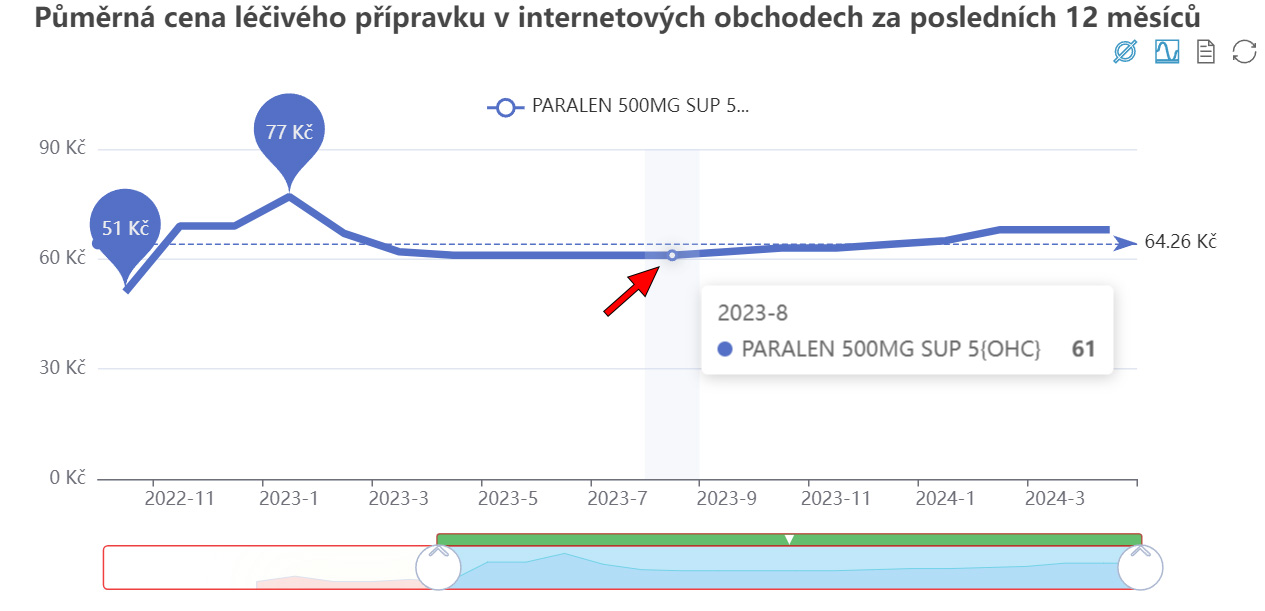
Zobrazení zdroje dat

Zobrazení grafu pro uživatele licence AISLP pro Windows
Při práci v AISLP s vybraným léčivým přípravkem si můžete v menu „Nástroje“ přímo zvolit možnost „Graf vývoje spotřeb“ nebo "Graf vývoje cen".
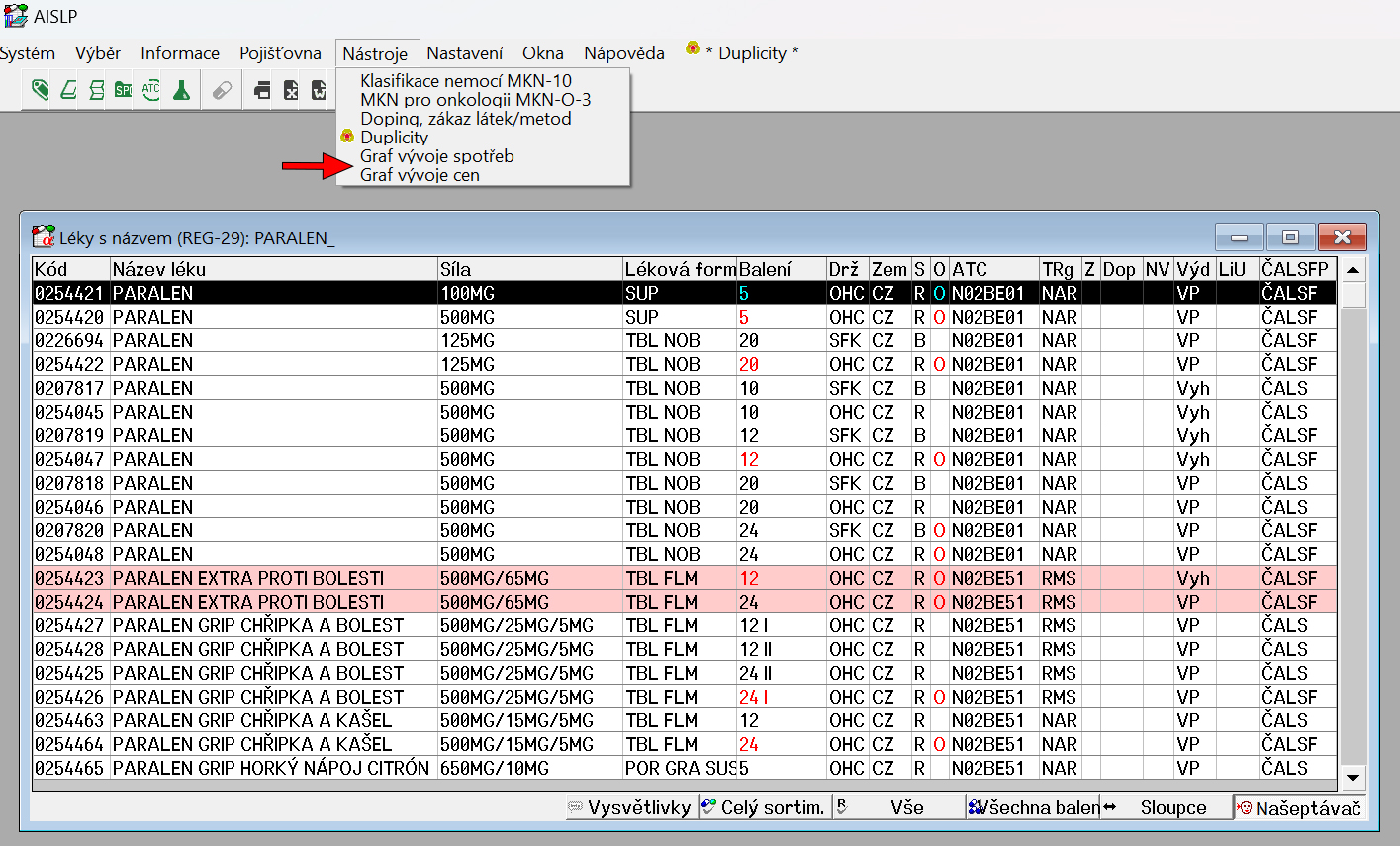
Další možností je po kliknutí v menu na "Nástroje" zvolit položku "Hlavní" a v okně informací o léčivém přípravku kliknout na příslušnou ikonku grafu.
Poté se Vám otevře nové okno prohlížeče se zobrazením grafů výdeje, nebo ceny léčivého přípravku, se kterým jste pracovali.
Zobrazení grafu pro uživatele internetové a intranetové verze AISLP
Na výpise léčivých přípravků klikněte na ikonku grafu
Vyberte graf, který chcete zobrazit. Poté se Vám otevře nové okno prohlížeče se zobrazením grafů výdeje, nebo ceny léčivého přípravku, se kterým jste pracovali.

Věříme, že se Vám tato aktualizace zalíbí a bude Vám k užitku při Vaší práci s programem AISLP.
Váš tým AISLP
www.aislp.cz
24.5.2024
Zobrazení grafů výdeje léčivých přípravků v internetové a intranetové verzi AISLP
Zobrazení grafů výdeje léčivých přípravků nyní i v internetové a intranetové verzi AISLP.
Vážení uživatelé AISLP,
zobrazení grafů výdeje léčivých přípravků, které jsme představili jako novinku pro uživatele Windows v září loňského roku, je nyní dostupné i pro internetovou a intranetovou verzi.
Tato aktualizace Vám umožní porovnat veškeré léčivé přípravky v dané ATC skupině.
Při práci s intranetovou nebo internetovou verzí AISLP kliknete ve výpisu LP na ikonku grafu. Otevře se vám detailní menu k vybranému LP s možností zobrazení konkrétního grafu. Kliknutím na tlačítko "Zobrazit graf" se vám otevře nové okno prohlížeče, ve kterém již můžete s vybraným grafem pracovat.
Viz. následující obrázky.
Výpis léčivých přípravků s ikonou grafu
Detailní menu vybraného léčivého přípravku
Zobrazení grafů a práce s nimi
Výběrem z rozbalovací nabídky si můžete přidat do grafického porovnání libovolné množství léčivých přípravků od různých držitelů z dané ATC skupiny.
Příklad na léku Paracetamol
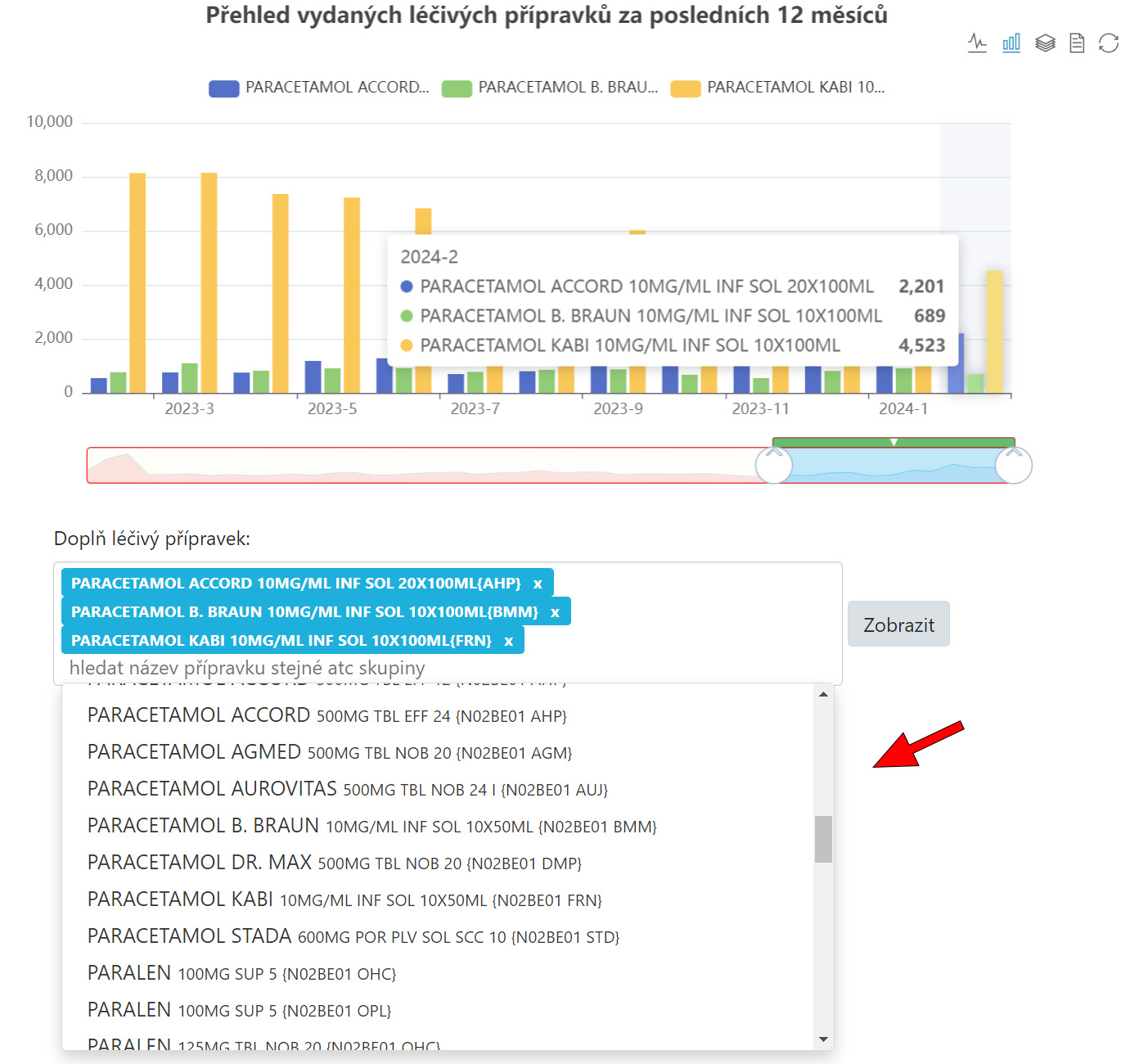
Léky se stejnou ATC skupinou
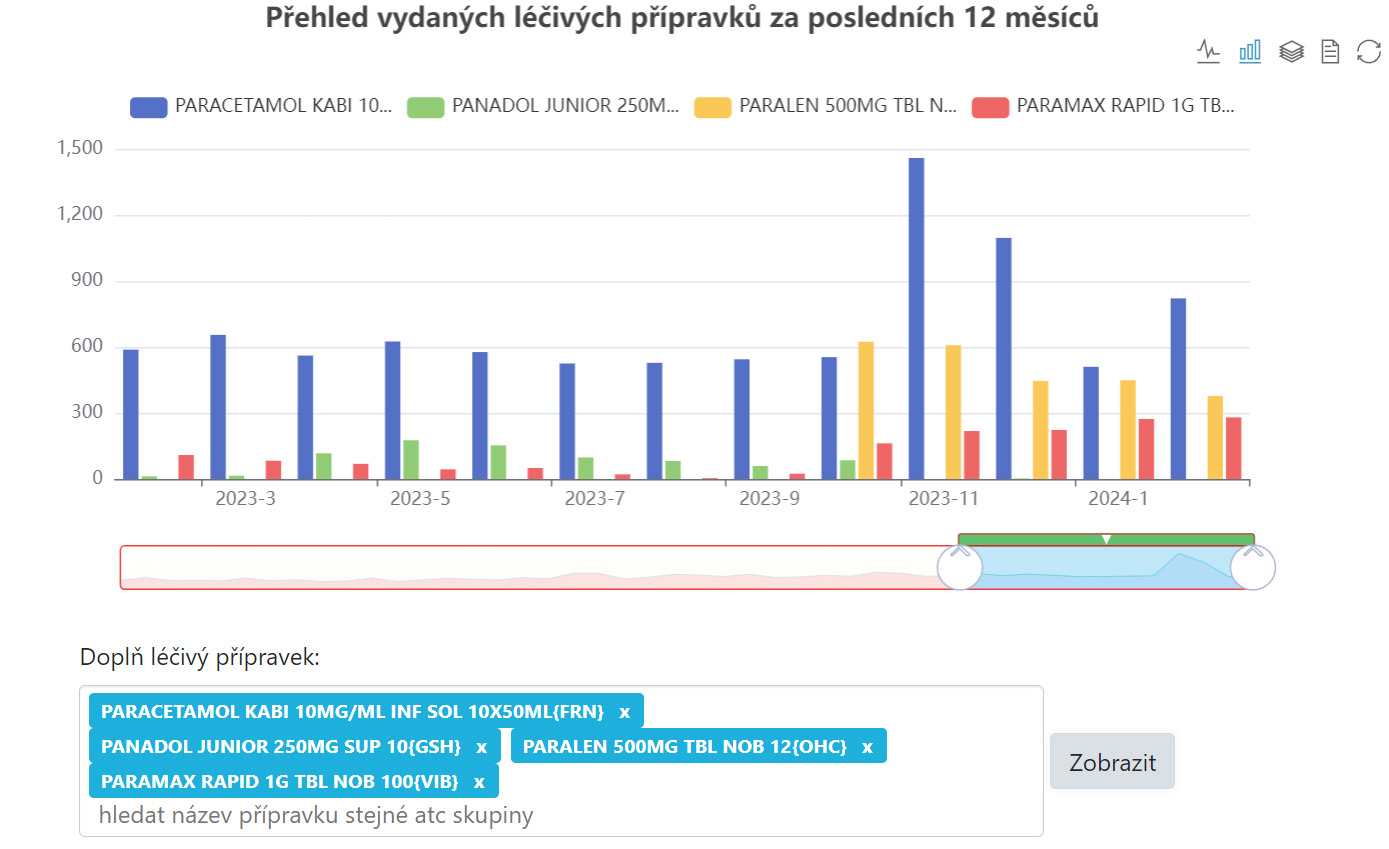
Dále v okně prohlížeče můžete využít i další možnosti, které Vám tento nástroj nabízí a které naleznete níže:
- volba formátu grafu a období zobrazení
- zobrazení výchozích dat
Stačí být držitelem platné licence AISLP a mít přístup k internetu. Ovládání je intuitivní a vyžaduje pouze základní znalost práce s podobnými datovými prohlížeči.
Níže naleznete několik ilustrativních obrázků ukazujících možnosti tohoto nástroje:
Volba jiného formátu grafu a období zobrazení
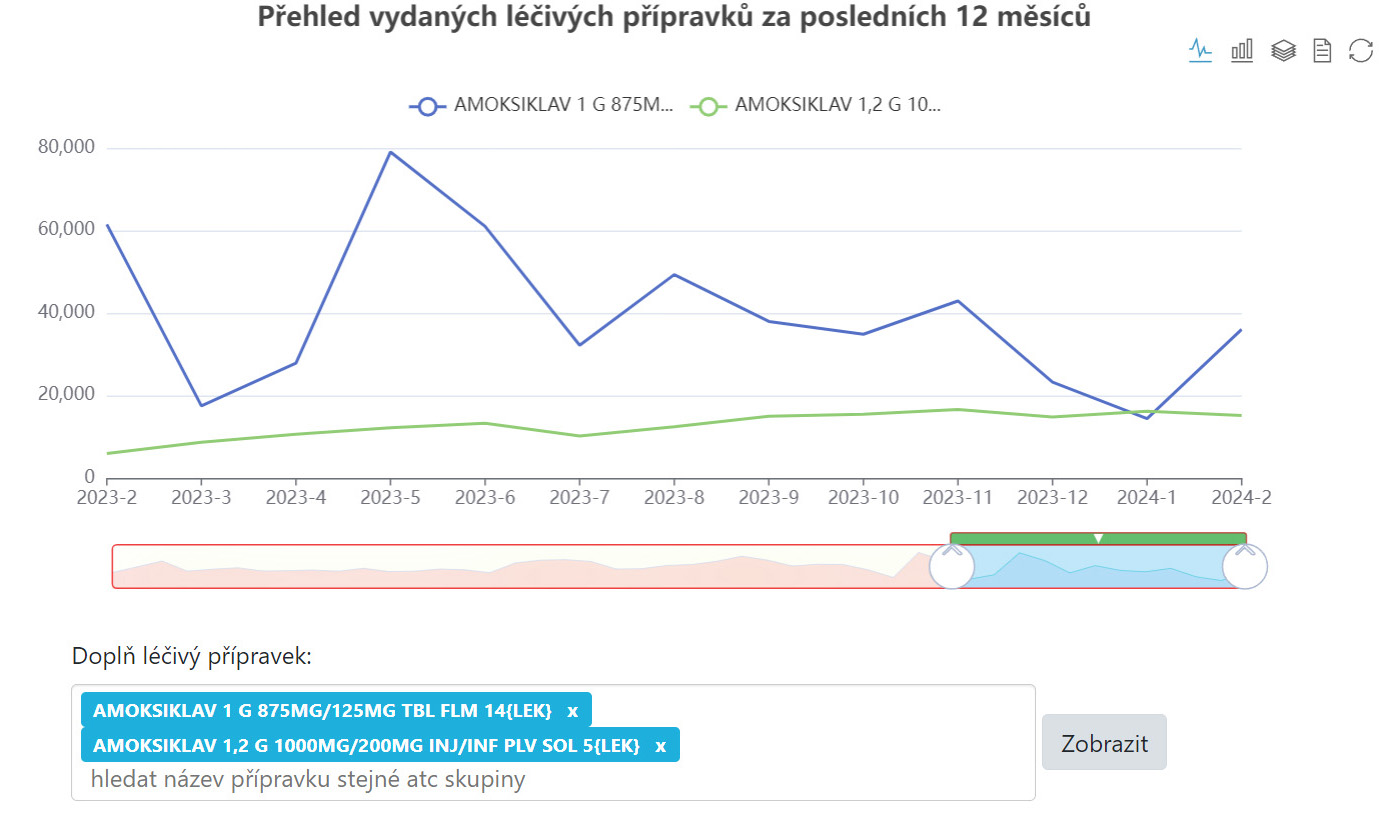
Zobrazení přehledů dat z LEK 13
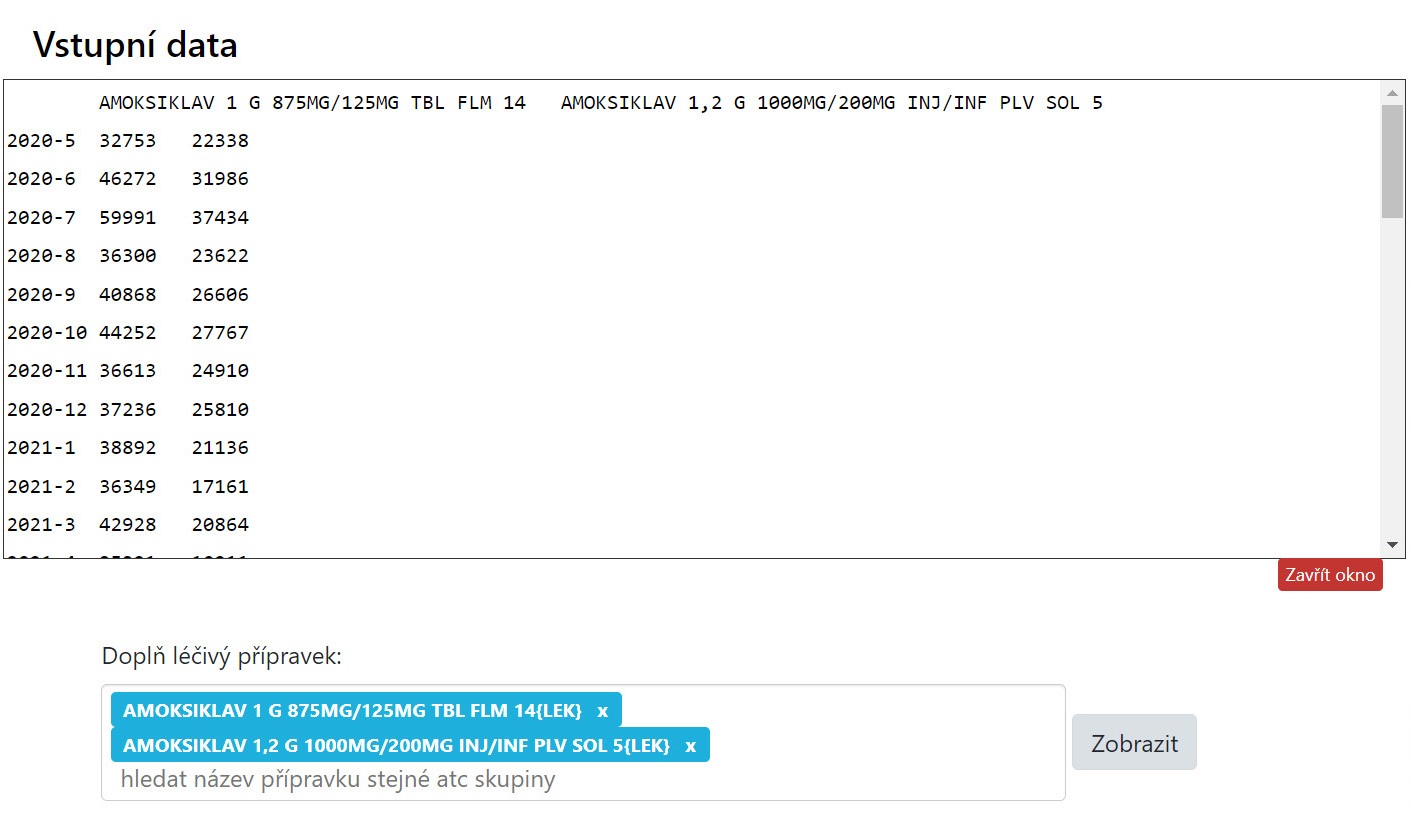
Přejeme Vám příjemnou práci a zábavu s tímto novým nástrojem!
Váš tým AISLP
www.aislp.cz
14.5.2024
Aktualizace KVĚTEN 2024
Aktualizace AISLP květen 2024 je připravena ke stažení.
Vážení uživatelé programu AISLP,
zasíláme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální květnové verzi AISLP. Připojujeme informaci o upozornění na režim výdeje RpM.
Nově praktická informace pro lékaře a lékárníky - režim výdeje RpM
U léčivých přípravků s obsahem vysoce návykových látek, jejichž výdej je vázán na eRecept s příznakem „modrý pruh“ (nebo na listinný recept s modrým pruhem), nově upozorňujeme na tuto skutečnost i v hlavičkách textů SmPC, PIL a Článku zkratkou režimu výdeje „RpM“.
Věříme, že pro Vás toto nové upozornění bude přínosné a užitečné.
Nově registrované léčivé přípravky/změny v registracích
Bimzelx 160 mg injekční roztok v předplněné injekční stříkačce/peru (bimekizumab) – rozšíření indikací: léčba aktivní středně těžké až těžké hidradenitis suppurativa (acne inversa) u dospělých při nedostatečné odpovědi na jinou konvenční systémovou léčbu HS.
Celldemic injekční suspenze v předplněné injekční stříkačce, vakcína proti zoonotické chřipce (H5N1), (povrchový antigen, inaktivovaná, obsahující adjuvans, připravovaná na buněčných kulturách) – k aktivní imunizaci proti subtypu H5N1 viru chřipky A u dospělých a dětí od 6 měsíců věku. Vakcína se má používat v souladu s oficiálními doporučeními.
Cibinqo 50 mg, 100 mg a 200 mg potahované tablety (abrocitinib) – rozšíření indikace: k léčbě středně závažné až závažné atopické dermatitidy u dospělých a dospívajících ve věku 12 let a starších, u kterých se zvažuje systémová léčba.
Efluelda Tetra injekční suspenze v předplněné injekční stříkačce – tetravalentní vakcína proti chřipce (štěpený virion, inaktivovaný), 60 mikrogramů HA/kmen – změna názvu přípravku.
Emblaveo 1,5 g/0,5 g prášek pro koncentrát pro infuzní roztok (aztreonam/avibaktam) – beta-laktamové antibiotikum ze skupiny monobaktamů a inhibitor beta-laktamáz – k léčbě infekcí u dospělých pacientů: komplikované intraabdominální infekce, nozokomiální pneumonie včetně ventilátorové pneumonie, komplikované infekce močových cest včetně pyelonefritidy. Přípravek je také indikován k léčbě infekcí vyvolaných aerobními gramnegativními mikroorganismy u dospělých pacientů s omezenými možnostmi léčby. Je třeba vzít v úvahu oficiální doporučené postupy pro správné používání antibakteriálních látek.
Epysqli 300 mg koncentrát pro infuzní roztok (ekulizumab) – rozšíření indikací: léčba atypického hemolyticko-uremického syndromu (aHUS) u dospělých a dětí.
Feiba 100 U/ml prášek a rozpouštědlo pro infuzní roztok (antiinhibiční komplex koagulačních faktorů) – nová síla.
Filspari 200 mg a 400 mg potahované tablety (sparsentan) – k léčbě dospělých s primární IgA nefropatií (IgAN) s proteinurií >= 1,0 g/den (nebo poměrem protein/kreatinin v moči >= 0,75 g/g).
Imfinzi 50 mg/ml koncentrát pro infuzní roztok (durvalumab) – cytostatikum, monoklonální protilátka – rozšíření indikace: monoterapie v první linii léčby dospělých pacientů s pokročilým nebo neresekovatelným hepatocelulárním karcinomem (HCC).
Incellipan injekční suspenze v předplněné injekční stříkačce, vakcína proti pandemické chřipce (H5N1), (povrchový antigen, inaktivovaná, obsahující adjuvans, připravovaná na buněčných kulturách) – k aktivní imunizaci proti chřipce v případě oficiálně vyhlášené pandemie. Vakcína se má používat v souladu s oficiálními doporučeními.
Kalydeco 13,4 mg granule v sáčku (ivakaftor) – nová síla pro děti od 1 měsíce s t.hm. od 3 kg k léčbě cystické fibrózy.
Mounjaro 2,5 mg/dávka, 5 mg/dávka, 7,5 mg/dávka, 10 mg/dávka, 12,5 mg/dávka a 15 mg/dávka KwikPen injekční roztok v předplněném peru (tirzepatid) – antidibetikum, GIP a GLP-1 agonista.
Diabetes mellitus 2.typu:
K léčbě dospělých s nedostatečně kontrolovaným diabetes mellitus 2. typu jako doplněk k dietě a cvičení: v monoterapii, pokud není podávání metforminu vhodné z důvodu nesnášenlivosti nebo kontraindikací; jako přídavná léčba k dalším léčivým přípravkům k léčbě diabetu.
Kontrola tělesné hmotnosti:
Jako doplněk k nízkokalorické stravě a zvýšené fyzické aktivitě pro kontrolu tělesné hmotnosti, včetně snížení a udržení tělesné hmotnosti u dospělých s počátečním indexem tělesné hmotnosti (BMI) >=30 kg/m2 (obezita) nebo >=27 kg/m2 až <30 kg/m2 (nadváha) za přítomnosti alespoň jednoho komorbidního stavu souvisejícího s hmotností (např. hypertenze, dyslipidemie, obstrukční spánková apnoe, kardiovaskulární onemocnění, prediabetes nebo diabetes mellitus 2. typu).
Palexia retard 25 mg, 50 mg, 100 mg, 150 mg, 200 mg a 250 mg (tapentadol) – rozšíření indikací na pediatrické pacienty: silné chronické bolesti u dospělých, dospívajících a dětí od 6 let, kterou adekvátně tlumí pouze opioidní analgetika.
Prevenar 20 injekční suspenze v předplněné injekční stříkačce, pneumokoková polysacharidová konjugovaná vakcína (20valentní, adsorbovaná) – změna názvu přípravku (dříve Apexxnar) a rozšíření indikace pro pediatrickou populaci: aktivní imunizace k prevenci invazivních onemocnění a pneumonie a akutní otitis media vyvolaných Streptococcus pneumoniae u kojenců, dětí a dospívajících ve věku od 6 týdnů do < 18 let.
Prevymis 240 mg a 480 mg koncentrát pro infuzní roztok
Prevymis 240 mg a 480 mg potahované tablety (letermovir) – rozšíření indikací: nově také k profylaxi rozvoje onemocnění způsobeného CMV u dospělých CMV séronegativních příjemců, kteří dostali transplantovanou ledvinu od CMV séropozitivního dárce [D+/R-].
Pyzchiva 130 mg koncentrát pro infuzní roztok (ustekinumab) – biosimilar – léčba dospělých pacientů se středně těžkou až těžkou aktivní Crohnovou chorobou, u kterých buď odpověď na konvenční terapii nebo na antagonistu tumor nekrotizujícího faktoru alfa (TNF-alfa) nebyla dostatečná nebo odezněla nebo tito pacienti netolerovali konvenční léčbu nebo léčbu TNF-alfa, případně jsou u nich tyto terapie kontraindikovány. Léčba dospělých pacientů se středně těžkou až těžkou aktivní ulcerózní kolitidou, u kterých buď odpověď na konvenční nebo biologickou léčbu nebyla dostatečná nebo došlo ke ztrátě odpovědi na léčbu nebo tito pacienti konvenční nebo biologickou léčbu netolerovali, případně je u nich tento způsob léčby kontraindikován.
Pyzchiva 45 mg a 90 mg injekční roztok v předplněné injekční stříkačce (ustekinumab) – biosimilar.
Plaková psoriáza: k léčbě středně těžké až těžké plakové psoriázy u dospělých, u kterých selhaly jiné systémové léčby, včetně podávání cyklosporinu, methotrexátu (MTX) a PUVA (psoralen a ultrafialové záření A), nebo kteří tyto léčby netolerují nebo jsou u nich kontraindikovány.
Plaková psoriáza u pediatrické populace: k léčbě středně těžké až těžké plakové psoriázy u dětí a dospívajících pacientů ve věku od 6 let, kteří nejsou dostatečně kontrolováni nebo netolerují jinou systémovou léčbu nebo fototerapii.
Psoriatická artritida (PsA): samostatně nebo v kombinaci s MTX k léčbě aktivní psoriatické artritidy u dospělých pacientů, pokud odpověď na předchozí léčbu nebiologickými chorobu modifikujícími antirevmatickými léky (DMARD) nebyla dostatečná.
Crohnova choroba: k léčbě dospělých pacientů se středně těžkou až těžkou aktivní Crohnovou chorobou, u kterých buď odpověď na konvenční terapii nebo na antagonistu tumor nekrotizujícího faktoru alfa (TNF-alfa) nebyla dostatečná nebo odezněla nebo tito pacienti netolerovali konvenční léčbu nebo léčbu TNF-alfa, případně jsou u nich tyto terapie kontraindikovány.
Ulcerózní kolitida: k léčbě dospělých pacientů se středně těžkou až těžkou aktivní ulcerózní kolitidou, u kterých buď odpověď na konvenční nebo biologickou léčbu nebyla dostatečná nebo došlo ke ztrátě odpovědi na léčbu nebo tito pacienti konvenční nebo biologickou léčbu netolerovali, případně je u nich tento způsob léčby kontraindikován.
Ryzneuta 20 mg injekční roztok (efbemalenograstim alfa) – zkrácení doby trvání neutropenie a snížení incidence febrilní neutropenie u dospělých pacientů léčených cytotoxickou chemoterapií pro maligní nádorové onemocnění (s výjimkou chronické myeloidní leukemie a myelodysplastického syndromu).
Tizveni 100 mg koncentrát pro infuzní roztok (tislelizumab) – cytostatikum, monoklonální protilátka - léčba nemalobuněčného karcinomu plic (NSCLC): v kombinaci s pemetrexedem a platinou nebo s karboplatinou a paklitaxelem nebo nab-paklitaxelem jako první linie léčby; v monoterapii k léčbě lokálně pokročilého nebo metastazujícího onemocnění.
Uptravi 100 mikrogramů potahované tablety (selexipag) – nová síla k dlouhodobé léčbě plicní arteriální hypertenze (PAH) u dospělých pacientů.
Voydeya 50 mg a 100 mg potahované tablety (danikopan) – přídatná léčba k ravulizumabu nebo ekulizumabu k léčbě dospělých pacientů s paroxysmální noční hemoglobinurií (PNH), kteří mají reziduální hemolytickou anémii.
Xromi 100 mg/ml perorální roztok (hydroxykarbamid) – cytostatikum – rozšíření indikace: prevence vazookluzivních komplikací srpkovité anémie u pacientů starších 9 měsíců.
Zoonotic Influenza Vaccine Seqirus injekční suspenze v předplněné injekční stříkačce,
vakcína proti zoonotické chřipce (H5N8) (povrchový antigen, inaktivovaná, s adjuvans), změna složení, aktualizace SmPC.
Zynys 500 mg koncentrát pro infuzní roztok (retifanlimab) – cytostatikum, monoklonální protilátka – monoterapie dospělých pacientů s metastazujícím nebo rekurentním lokálně pokročilým karcinomem z Merkelových buněk (MCC), u kterého nelze provést chirurgický zákrok ani radioterapii.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
15.4.2024
Aktualizace DUBEN 2024
Aktualizace AISLP duben 2024 je připravena ke stažení.
Vážení uživatelé programu AISLP,
zasíláme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální dubnové verzi AISLP.
Emoxen Plus 500 mg/20 mg tablety s řízeným uvolňováním (naproxen, esomeprazol) – nová fixní kombinace nesteroidního antiflogistika a inhibitoru protonové pumpy – symptomatická léčba osteoartrózy, revmatoidní artritidy a ankylozující spondylitidy u dospělých pacientů s rizikem rozvoje žaludečních a/nebo duodenálních vředů při podávání nesteroidních antirevmatik (NSAID), u kterých léčba nižšími dávkami naproxenu nebo jinými NSAID není dostatečně účinná.
Exblifep 2 g/0,5 g prášek pro koncentrát pro infuzní roztok (cefepim/enmetazobaktam) – cefepim a inhibitor beta-laktamáz – k léčbě následujících infekcí u dospělých: komplikované infekce močových cest (cUTI), včetně pyelonefritidy; nozokomiální pneumonie (HAP), včetně ventilátorové pneumonie (VAP). Léčba pacientů s bakteriemií, která se vyskytuje v souvislosti s některou z uvedených infekcí nebo u níž existuje podezření na tuto souvislost. Je třeba vzít v úvahu oficiální doporučení týkající se vhodného používání antibakteriálních látek.
Foclivia injekční suspenze/injekční suspenze v předplněné injekční stříkačce, vakcína proti pandemické chřipce (H5N1) (inaktivovaný povrchový antigen, s adjuvans) – rozšíření indikací o pediatrickou populaci: nově k očkování jedinců od 6 měsíců věku.
Frimig Duo 85 mg/500 mg potahované tablety (sumatriptan, naproxen) – nová fixní kombinace triptanu a nesteroidního antiflogistika – akutní léčba bolesti hlavy při záchvatech migrény s aurou nebo bez aury u dospělých pacientů, u kterých je léčba sumatriptanem nedostatečná.
Fynzur 2,275 mg/ml kožní sprej, roztok (finasterid) – dermatologikum – k lokální léčbě dospělých mužů ve věku od 18 do 41 let s mírnou až střední mužskou alopecií (androgenní alopecie) k zesílení růstu vlasů a k zabránění dalšímu vypadávání vlasů.
Pliaglis 70 mg + 70 mg krém (lidokain, tetrakain) – k navození lokální kožní anestezie na neporušené kůži před dermatologickými zákroky u dospělých.
Tresiba 100 jednotek/ml injekční roztok v předplněném peru/v zásobní vložce
Tresiba 200 jednotek/ml injekční roztok v předplněném peru (insulin-degludek) – dlouhodobě působící inzulinový analog – aktualizace bodu 4.6 a 5.1 SmPC: insulin-degludek může být v klinicky indikovaných případech podáván u těhotných žen.
VeraSeal roztoky pro tkáňové lepidlo (lidský fibrinogen, lidský trombin) – rozšíření indikace pro všechny věkové skupiny.
Zinplava 25 mg/ml koncentrát pro infuzní roztok (bezlotoxumab) – antibakteriální monoklonální protilátka – rozšíření indikací o pediatrickou populaci: k prevenci rekurence infekce vyvolané bakterií Clostridioides difficile (CDI) u dospělých a pediatrických pacientů ve věku 1 rok a starších, kteří jsou rekurencí CDI vysoce ohroženi.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
3.4.2024
Zobrazení grafů výdeje léčivých přípravků
Novinky v zobrazení grafů výdeje léčivých přípravků.
Vážení uživatelé AISLP,
pro ty z Vás, kteří si oblíbili práci s grafy, nebo naopak ještě tuto možnost neobjevili, přinášíme aktualizaci týkající se možnosti grafického zobrazení výdeje léčivých přípravků. Tato aktualizace Vám umožní porovnat veškeré léčivé přípravky v dané ATC skupině.
Nově si můžete výběrem z rozbalovací nabídky přidat do grafického porovnání libovolné množství léčivých přípravků od různých držitelů z dané ATC skupiny.
Viz. příklady a detailnější popis níže.
Při práci v AISLP s vybraným konkrétním LP můžete v menu „Nástroje“ zvolit možnost „Statistika, Grafy“ nebo při zobrazení hlavní informace o LP kliknout na tlačítko „Grafy“. Tím se Vám otevře nové okno prohlížeče se zobrazením výdejů LP, se kterým jste pracovali.
Příklad na léku Paracetamol
Léky se stejnou ATC skupinou
Dále v okně prohlížeče můžete využít i další možnosti, které Vám tento nástroj nabízí a které naleznete níže:
- volba formátu grafu a období zobrazení
- zobrazení výchozích dat
Stačí být držitelem platné licence Windows AISLP a mít přístup k internetu. Ovládání je intuitivní a vyžaduje pouze základní znalost práce s podobnými datovými prohlížeči.
Níže naleznete několik ilustrativních obrázků ukazujících možnosti tohoto nástroje:
Volba jiného formátu grafu a období zobrazení
Zobrazení přehledů dat z LEK 13
Přejeme Vám příjemnou práci a zábavu s tímto novým nástrojem!
Váš tým AISLP
www.aislp.cz
22.3.2024
Velikonoční prodejní akce
Máte zájem o rozšíření své licence AISLP?

Více informací na www.aislp.cz, kliknutím na reklamní proužek.
Váš tým AISLP
www.aislp.cz
19.3.2024
SÚKL informuje o stažení uvedených šarží léčivého přípravku Methylrosanilinii chloridi solutio 0,5%, 10g až z úrovně zdravotnických zařízení.
15.3.2024
Aktualizace BŘEZEN 2024
Aktualizace AISLP březen 2024 je připravena ke stažení.
Vážení uživatelé programu AISLP,
zasíláme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální březnové verzi AISLP.
Ayvakyt 25, 50, 100, 200 a 300 mg potahované tablety (avapritinib) – cytostatikum, inhibitor proteinkináz – rozšíření indikace: léčba dospělých pacientů s indolentní systémovou mastocytózou (ISM) se středně těžkými až těžkými symptomy, které nejsou dostatečně kontrolovány symptomatickou léčbou.
Brukinsa 80 mg tvrdé tobolky (zanubrutinib) – cytostatikum, inhibitor proteinkináz – rozšíření indikace: v kombinaci s obinutuzumabem k léčbě dospělých pacientů s refrakterním nebo relabovaným folikulárním lymfomem (FL), kteří podstoupili alespoň 2 předchozí systémové léčby.
Casgevy 4-13 milionů buněk infuzní disperze (exagamglogen autotemcel) – přípravek buněčné léčby ex vivo, léčba beta-talasemie závislé na transfuzi a léčba těžké srpkovité anémie s opakujícími se vazookluzivními krizemi u pacientů od 12 let, u nichž je vhodná transplantace hematopoetických kmenových buněk (HSCT) a není k dispozici příbuzný dárce HSC s odpovídajícím HLA.
Empagliflozin Polpharma 10 mg a 25 mg potahované tablety – antidiabetikum, inhibitor SGLT2 – první generikum s léčivou látkou empagliflozin: k léčbě dospělých pacientů s nedostatečně kontrolovaným diabetes mellitus 2. typu jako přídavná terapie k dietě a tělesnému cvičení: jako monoterapie, když je použití metforminu nevhodné z důvodu nesnášenlivosti; v kombinaci s jinými léčivými přípravky k léčbě diabetu. K léčbě dospělých pacientů se symptomatickým chronickým srdečním selháním.
Enhertu 100 mg prášek pro koncentrát pro infuzní roztok (trastuzumab deruxtekan) – cytostatikum, konjugát monoklonální protilátky a cytostatika – rozšíření indikace: monoterapie dospělých pacientů s pokročilým NSCLC, jejichž nádory mají aktivační mutaci HER2 (ERBB2) a jejichž stav vyžaduje systémovou léčbu po chemoterapii na bázi platiny s imunoterapií nebo bez ní.
Ibuprofen Gen.Orph. 5 mg/ml injekční roztok (ibuprofen) – léčba hemodynamicky významného otevřeného ductus arteriosus u předčasně narozených novorozenců mladších než 34 týdnů gestačního věku.
Jemperli 500 mg koncentrát pro infuzní roztok (dostarlimab) – cytostatikum, monoklonální protilátka – rozšíření indikace: v kombinaci s karboplatinou a paklitaxelem k léčbě dospělých pacientek s primárním pokročilým nebo rekurentním karcinomem endometria s deficitní opravou chybného párování bází (dMMR) či vysokou mikrosatelitovou nestabilitou (MSI-H), které jsou indikované k systémové léčbě.
Livizux 20 mg, 30 mg, 40 mg, 50 mg, 60 mg a 70 mg tvrdé tobolky (lisdexamfetamin) – součást komplexního léčebného programu při poruše pozornosti s hyperaktivitou (ADHD) u dětí od 6 let, pokud se odpověď na předchozí léčbu methylfenidátem považuje za klinicky neodpovídající.
Mounjaro 2,5 mg, 5 mg, 7,5 mg, 10 mg, 12,5 mg a15 mg injekční roztok v předplněném peru/ v injekční lahvičce (tirzepatid) – antidibetikum, GIP a GLP-1 agonista – rozšíření indikací:
Diabetes mellitus 2.typu:
K léčbě dospělých s nedostatečně kontrolovaným diabetes mellitus 2. typu jako doplněk k dietě a cvičení: v monoterapii, pokud není podávání metforminu vhodné z důvodu nesnášenlivosti nebo kontraindikací; jako přídavná léčba k dalším léčivým přípravkům k léčbě diabetu.
Kontrola tělesné hmotnosti:
Jako doplněk k nízkokalorické stravě a zvýšené fyzické aktivitě pro kontrolu tělesné hmotnosti, včetně snížení a udržení tělesné hmotnosti u dospělých s počátečním indexem tělesné hmotnosti (BMI)
>=30 kg/m2 (obezita) nebo >=27 kg/m2 až <30 kg/m2 (nadváha) za přítomnosti alespoň jednoho komorbidního stavu souvisejícího s hmotností (např. hypertenze, dyslipidemie, obstrukční spánková apnoe, kardiovaskulární onemocnění, prediabetes nebo diabetes mellitus 2. typu).
Ryeqo 140 mg/1 mg/0,5 mg potahované tablety (relugolix, hemihydrát estradiolu, norethisteron-acetát) – gynekologikum – rozšíření indikace: symptomatická léčba endometriózy u žen s předchozí medikamentózní nebo chirurgickou léčbou endometriózy v anamnéze.
Skyclaris 50 mg tvrdé tobolky (omaveloxolon) – léčba Friedreichovy ataxie u dospělých a dospívajících ve věku od 16 let.
Velsipity 2 mg potahované tablety (etrasimod) – léčba pacientů ve věku 16 let a starších se středně těžkou až těžkou aktivní ulcerózní kolitidou (UC), kteří měli nedostatečnou odpověď, ztrátu odpovědi nebo intoleranci buď na konvenční léčbu, nebo na biologickou léčivou látku.
Voxzogo 0,4 mg, 0,56 mg a 1,2 mg prášek a rozpouštědlo pro injekční roztok (vosoritid) – rozšíření indikace na pacienty od 4 měsíců: léčba achondroplazie u pacientů ve věku od 4 měsíců, jejichž epifýzy nejsou uzavřené. Diagnóza achondroplazie má být potvrzena vhodným genetickým vyšetřením.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
www.aislp.cz
14.3.2024
SÚKL byl upozorněn na několik případů závažného poškození oka po podání nitrooční injekce afliberceptu.
8.3.2024
Informace o závadách v jakosti, opatřeních z registračních důvodů a nežádoucích účincích léčiv, padělcích a nelegálních přípravcích.
7.3.2024
Z důvodu odstávky nebude dne 08. 03. 2024 od 20 hodin do 11.3.2024 cca 05:00 hodin bude probíhat odstávka DC B z důvodu technologického upgrade.
Ve dnech 8.3.2024 od 20:00 hodin do 11.3.2024 cca 05:00 hodin bude probíhat odstávka DC B z důvodu technologického upgrade. Po dobu odstávky nebudou funkční následující aplikace a prostředí:
HSZ – Hlášení skladových zásob,
LEK13 – Hlášení výdeje v lékárnách,
HOPL – Hlášení opiátů,
DIS13 – Hlášení dodávek distributorů LP,
REG13 – Hlášení dodávek držitelů LP,
HZ – Závady v jakosti,
KLP – Hlášení konopí,
MAH11r – Žádost o výjimku z ochranných prvků LP,
NU – Hlášení nežádoucích účinků - formulář na webu,
záložní lokalita systémů eRecept, ePoukaz, ISZP apod.,
testovací prostředí všech aplikací,
7.3.2024
Sekce regulace zdravotnických prostředků informuje o spuštění Informačního systému zdravotnických prostředků (dále jen “ISZP”) dle § 7 zákona č. 375/2022 Sb., o zdravotnických prostředcích a diagnostických zdravotnických prostředcích in vitro (dále jen „zákon o prostředcích“).
6.3.2024
Z důvodu odstávky nebude dne 08. 03. 2024 od 20 hodin do 11.3.2024 cca 05:00 hodin bude probíhat odstávka DC B z důvodu technologického upgrade.
Ve dnech 8.3.2024 od 20:00 hodin do 11.3.2024 cca 05:00 hodin bude probíhat odstávka DC B z důvodu technologického upgrade. Po dobu odstávky nebudou funkční následující aplikace a prostředí:
HSZ – Hlášení skladových zásob,
LEK13 – Hlášení výdeje v lékárnách,
HOPL – Hlášení opiátů,
DIS13 – Hlášení dodávek distributorů LP,
REG13 – Hlášení dodávek držitelů LP,
HZ – Závady v jakosti,
KLP – Hlášení konopí,
MAH11r – Žádost o výjimku z ochranných prvků LP,
NU – Hlášení nežádoucích účinků - formulář na webu,
záložní lokalita systémů eRecept, ePoukaz, ISZP apod.,
testovací prostředí všech aplikací,
5.3.2024
Státní ústav pro kontrolu léčiv (SÚKL) zaznamenal na českém trhu zvýšený výskyt výrobků obsahujících účinnou látku N-acetylcystein (syn. acetylcystein, N-acetyl-L-cystein).
1.3.2024
Státní ústav pro kontrolu léčiv informuje o závadě v jakosti léčivého přípravku ACIDI BORICI AQUA OPHTHALMICA CUM T. Ten se používá při očních potížích, jako jsou např. lehčí formy zánětů spojivek, dále při pálení, řezání a svědění oka způsobené zevními vlivy.
28.2.2024
Filtrování v internetové a intranetové verzi AISLP II.
Používáte již filtrování v aplikaci AISLP? Jaké možnosti Vám tato nová funkce přináší?
Vážení uživatelé programu AISLP,
s funkcí filtrování léčivých přípravků/látek jsme Vás seznámili v předchozí novince. Nyní Vám chceme ukázat jak jednoduše s filtry pracovat a jaké možnosti tato funkce má.
Zde je jednoduchý příklad vyhledání a filtrování na léčivém přípravku Nalgesin. První obrázek ukazuje základní vyhledání tohoto přípravku a dále následují ukázky použití jednotlivých filtrů.
Základní vyhledání přípravku Nalgesin
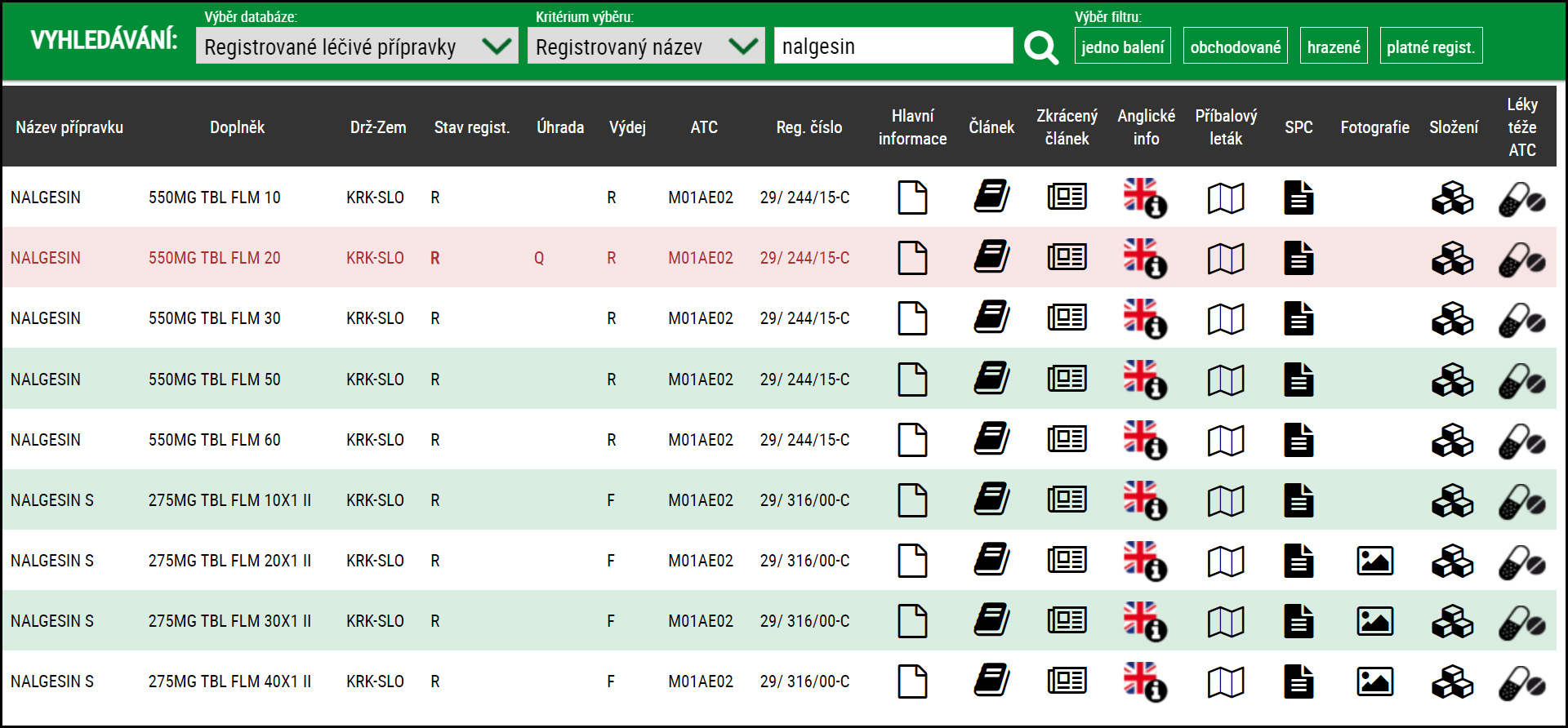
Použitý filtr "jedno balení":
Tento filtr rychle skryje všechna další balení pro kombinaci Název - Síla - Léková forma. Ponechá jednoho zástupce, zpravidla takového, který je hrazen / obchodován, aktuálně registrován a nejmenšího balení.
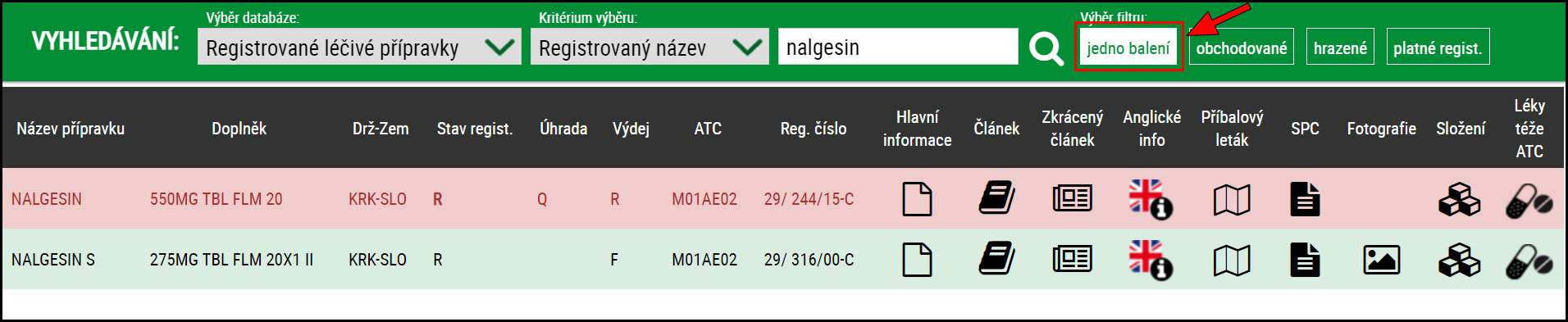
Použitý filtr "obchodované":
Filtr zobrazí přípravky, které byly dodávány do lékáren a zdravotnických zařízení v posledních 6 měsících.
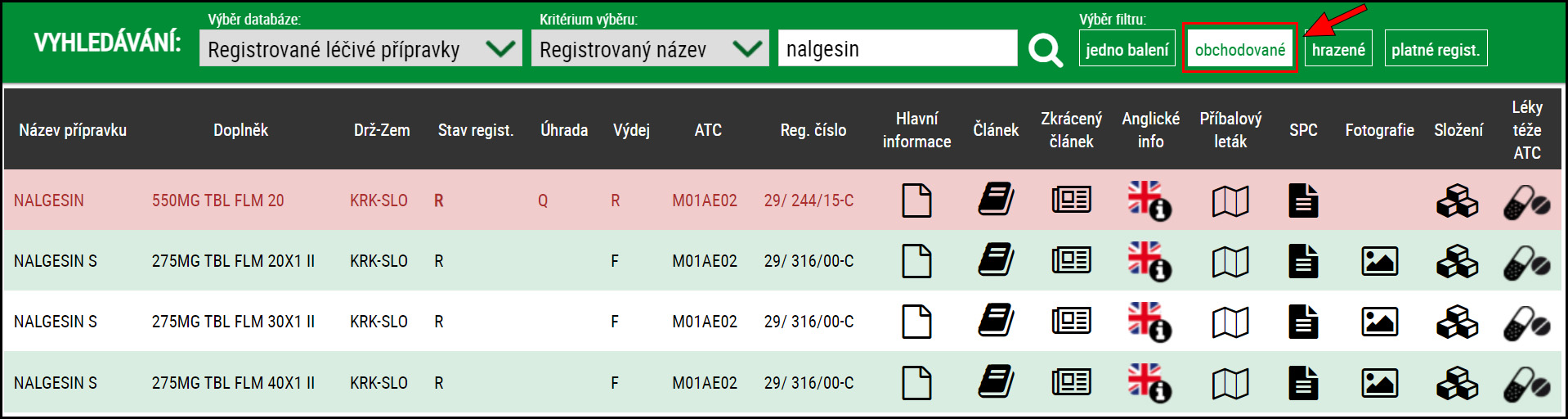
Použitý filtr "hrazené":
Tento filtr zobrazí přípravky se stanovenou úhradou v číselnících SZP (Svaz zdravotních pojišťoven).
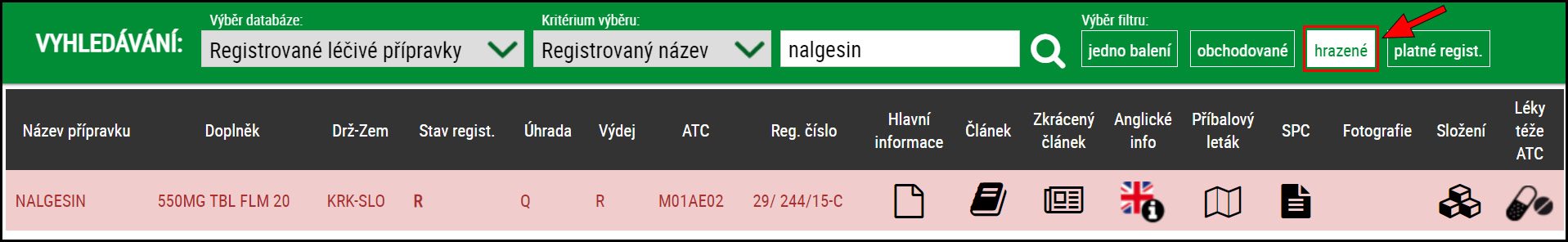
Použitý filtr "platné registrace":
Při použití tohoto filtru uvidíte jen přípravky s platnou registrací (včetně kódů, případně i názvů před změnou platné registrace - nejčastěji změna držitele).
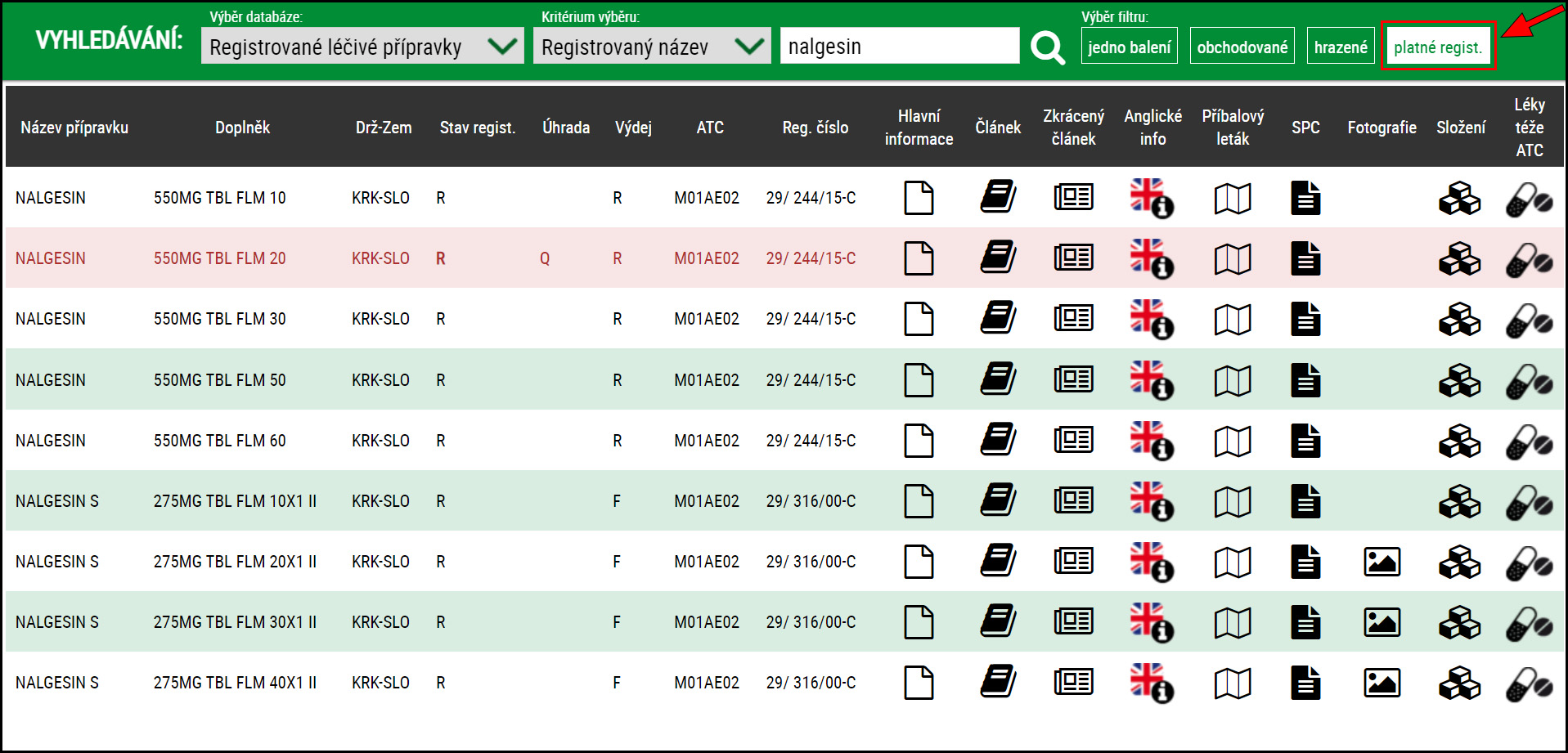
Přejeme Vám hodně spokojenosti při používání programu AISLP a těšíme se na Vaší případnou zpětnou vazbu.
Váš tým AISLP
www.aislp.cz
info@aislp.cz
19.2.2024
Filtrování v internetové a intranetové verzi AISLP
Verze intranet a internet nyní včetně filtrů.
Vážení uživatelé programu AISLP,
rádi bychom Vám představili novinku, kterou jsme pro Vás aktuálně připravili. Jedná se o filtrování léčivých přípravků/látek v internetové a intranetové verzi AISLP. Zprovozněním této funkce dále pokračujeme ve sbližování podoby i práce s AISLP v různých verzích a zároveň jsme tím doplnili hodně poptávanou funkcionalitu ze strany uživatelů v této verzi AISLP, a tou je možnost použití "Filtrů".
Tyto filtry Vám maximálně zpřehlední pohled na dlouhé seznamy léčivých přípravků a látek. Zredukují záznamy tak, aby se Vám požadované výpisy lépe prohlížely a mohli jste se tak rychleji zorientovat. I z naší zkušenosti je to jedna z nejvíce využívaných funkčních "vychytávek", která doposud byla k dispozici uživatelům ve verzi pro Windows.
Filtry naleznete ihned na úvodní stránce.
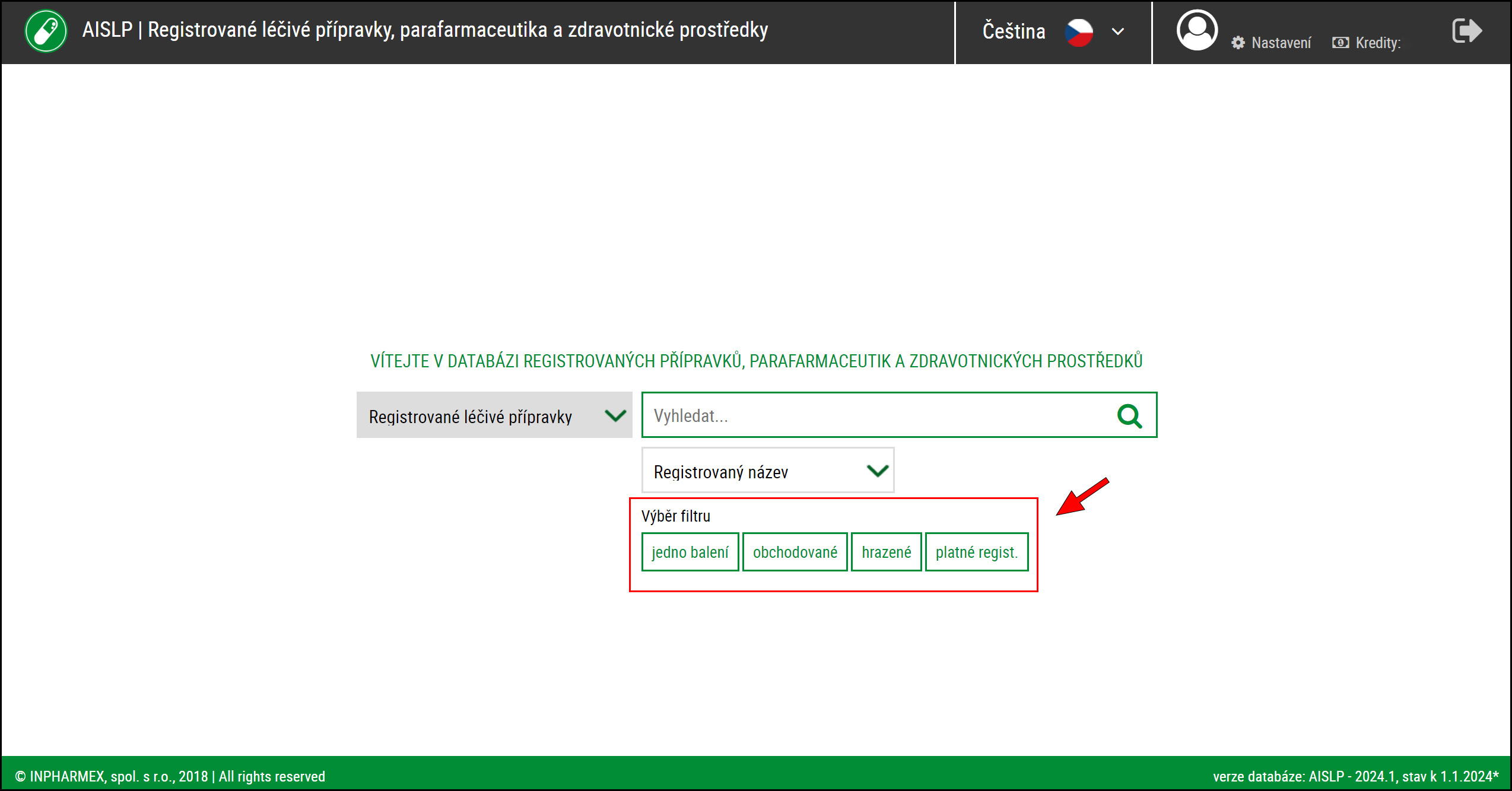
Další možnost filtrování máte na výpisu léčivých přípravků/látek.
Filtry mezi sebou můžete jakkoliv kombinovat, abyste nalezli přesně takový výsledek, který potřebujete. Zapnutí a vypnutí je čistě intuitivní a záleží na Vaší preferenci zobrazení dat. Pro ty z Vás, kteří používají intranetovou/internetovou verzi AISLP a zaujala Vás možnost využití filtrů, připravujeme detailnější seznámení s prací s filtry.
Doufáme, že vám tato novinka ještě více usnadní práci s programem AISLP.
Váš tým AISLP
www.aislp.cz
15.2.2024
SÚKL informuje o stažení uvedených šarží léčivého přípravku Fentanyl-Ratiopharm, 12mcg/h tdr. emp. 5X2,1mg až z úrovně zdravotnických zařízení.
14.2.2024
Aktualizace ÚNOR 2024
Aktualizace AISLP únor 2024 je připravena ke stažení.
Vážení uživatelé programu AISLP,
zasíláme Vám výběr z nově registrovaných léčivých přípravků (léčivých látek) a ze změn v registracích, které naleznete v aktuální únorové verzi AISLP.
Jardiance 10 mg a 25 mg potahované tablety (empagliflozin) – antidiabetikum, inhibitor SGLT2 – rozšíření indikací o pediatrickou populaci: k léčbě dospělých a dětí ve věku 10 let a starších s nedostatečně kontrolovaným diabetes mellitus 2. typu jako přídavná terapie k dietě a tělesnému cvičení: jako monoterapie, když je použití metforminu nevhodné z důvodu nesnášenlivosti; v kombinaci s jinými léčivými přípravky k léčbě diabetu.
Krazati 200 mg potahované tablety (adagrasib) – cytostatikum – monoterapie dospělých pacientů s pokročilým nemalobuněčným karcinomem plic (NSCLC) s mutací KRAS G12C a progresí onemocnění po nejméně jedné předchozí systémové léčbě.
Metalyse 5000 jednotek prášek pro injekční roztok (tenekteplasa) – trombolytická léčba u dospělých pacientů s akutní ischemickou cévní mozkovou příhodou do 4,5 hodiny od okamžiku, kdy byl pacient naposledy bez příznaků, a po vyloučení intrakraniálního krvácení.
NexoBrid 2 g a 5 g prášek a gel pro přípravu gelu (bromelainy) – proteolytické enzymy – rozšíření indikací o pediatrickou populaci: přípravek je indikován u všech věkových skupin k odstranění eschary (popáleninového příškvaru) u pacientů s termickými hlubokými popáleninami stupně IIb (deep partial-thickness burn) a III (full-thickness burn).
Omjjara 100 mg, 150 mg, 200 mg potahované tablety (momelotinib) – cytostatikum, inhibitor proteinkináz – léčba splenomegalie nebo příznaků souvisejících s onemocněním u dospělých pacientů se středně závažnou až závažnou anémií, kteří mají primární myelofibrózu, myelofibrózu po polycythaemia vera nebo myelofibrózu po esenciální trombocytemii, a kteří dosud nebyli léčeni inhibitorem Janusových kináz (JAK) nebo byli léčeni ruxolitinibem.
Praluent 75 mg a 150 mg injekční roztok v předplněném peru
Praluent 75 mg, 150 mg a 300 mg injekční roztok v předplněné injekční stříkačce (alirokumab) – rozšíření indikací o pediatrickou populaci: přípravek je indikován k léčbě dospělých pacientů s primární hypercholesterolemií (heterozygotní familiární a nefamiliární) nebo smíšenou dyslipidemií a pediatrických pacientů ve věku od 8 let s heterozygotní familiární hypercholesterolemií (HeFH) jako doplněk k dietním opatřením:
- v kombinaci se statinem nebo se statinem a další hypolipidemickou léčbou u pacientů, u kterých nelze dostáhnout cílových hodnot LDL-cholesterolu (LDL-C) maximální tolerovanou dávkou statinů, nebo
- samostatně nebo v kombinaci s další hypolipidemickou léčbou u pacientů, kteří netolerují statiny nebo, u kterých je podávání statinů kontraindikováno.
Rezzayo 200 mg prášek pro koncentrát pro infuzní roztok (rezafungin-acetát) – amtimykotikum – k léčbě invazivní kandidózy u dospělých. Je zapotřebí vzít v úvahu oficiální pokyny ke správnému použití antimykotik.
Rystiggo 140 mg/ml injekční roztok (rozanolixizumab) – doplněk ke standardní léčbě generalizovaného onemocnění myasthenia gravis (gMG) u dospělých pacientů, kteří jsou pozitivní na protilátky proti acetylcholinovému receptoru (AChR) nebo protilátky proti svalově specifické tyrozin-kináze (MuSK).
Spexotras 0,05 mg/ml prášek pro perorální roztok (trametinib) – cytostatikum, inhibitor proteinkináz – v kombinaci s dabrafenibem k léčbě dětí od 1 roku s gliomem nízkého stupně (LGG) nebo vysokého stupně (HGG) s mutací V600E genu BRAF.
Skyrizi 90 mg injekční roztok (risankizumab) – nová síla přípravku: k léčbě dospělých pacientů se středně těžkou až těžkou aktivní Crohnovou chorobou s nedostatečnou odpovědí, se ztrátou odpovědi nebo intolerancí konvenční léčby nebo biologické léčby.
Tolvecamo 40 mg/5 mg/12,5 mg, 80 mg/5 mg/12,5 mg, 80 mg/10 mg/12,5 mg tablety a 80 mg/10 mg/25 mg tablety (telmisartan, amlodipin-besilát, hydrochlorothiazid) – přípravek je indikován jako náhrada léčby u dospělých pacientů s esenciální hypertenzí, jejichž krevní tlak je odpovídajícím způsobem kontrolován dvojkombinací telmisartanu a hydrochlorothiazidu a monokomponentním přípravkem s obsahem amlodipinu podávanými souběžně ve stejných dávkách jako v kombinaci, ale jako samostatné tablety.
Uzpruvo 45 mg a 90 mg injekční roztok v předplněné injekční stříkačce (ustekinumab) – biosimilar.
Plaková psoriáza: k léčbě středně těžké až těžké plakové psoriázy u dospělých, u kterých selhaly jiné systémové léčby, včetně podávání cyklosporinu, methotrexátu (MTX) a PUVA (psoralen a ultrafialové záření A), nebo kteří tyto léčby nesnášejí nebo jsou u nich kontraindikovány.
Plaková psoriáza u pediatrické populace: k léčbě středně těžké až těžké plakové psoriázy u dětí a dospívajících pacientů ve věku od 6 let a starších, kteří nejsou pod dostatečnou kontrolou nebo nesnášejí jiné systémové léčby nebo fototerapii.
Psoriatická artritida (PsA): samostatně nebo v kombinaci s MTX k léčbě aktivní psoriatické artritidy u dospělých pacientů, pokud odpověď na předchozí léčbu nebiologickými, onemocnění modifikujícími antirevmatickými přípravky (DMARD) nebyla dostatečná.
Crohnova choroba: k léčbě dospělých pacientů se středně těžkou až těžkou aktivní Crohnovou chorobou, u kterých buď odpověď na konvenční terapii nebo na antagonistu tumor nekrotizujícího faktoru alfa (TNF-alfa) nebyla dostatečná nebo odezněla nebo tito pacienti netolerovali konvenční léčbu nebo léčbu TNF-alfa, případně jsou u nich tyto terapie kontraindikovány.
Přejeme Vám hodně spokojenosti při používání programu AISLP.
Váš tým AISLP
7.2.2024
Informace o závadách v jakosti, opatřeních z registračních důvodů a nežádoucích účincích léčiv, padělcích a nelegálních přípravcích.
7.2.2024
SÚKL informuje o stažení uvedené šarže léčivého přípravku Metalcaptase, 300mg tbl. ent. 50 až z úrovně zdravotnických zařízení.